1.本发明属于功能电磁材料技术领域,具体涉及一种低频及高频电磁波吸收剂及其制备方法。
背景技术:
2.无线通讯设备和电子设备的快速发展引发了大量的微波辐射,对生存环境产生信号污染和干扰及对人类生存健康产生无法预测的潜在危害,亟待开发先进电磁波吸收材料解决并匹配面临的技术问题。于此同时,新一代移动通信技术的快速更替更是加速了对先进电磁波吸收材料的迫切需求,需要开发满足“薄、轻、宽、强”的微波吸收材料。碳基材料因其轻质、强介电损耗、高电导率等优点,在实现高性能电磁波衰减方面得到了广泛的青睐。在目前的研究中,将碳材料(还原氧化石墨烯、碳纳米管、碳纤维、胶态碳等)与磁性材料(磁性金属、铁氧体、磁性氧化物)相结合,被认为是拓宽电磁波吸收剂有效带宽的有效途径。例如:氧化石墨烯与氧化铁、碳纳米管与氧化铁、氧化石墨烯与钴镍合金等一系列优化的复合型微波吸收剂被报道。此外,电磁波吸收剂的微观结构对电磁波吸收的阻抗变化、吸波性能影响明显。在以往的报道中,微观形貌的调控仅局限于金属及其金属氧化物微观形貌变化对电磁波吸收性能的影响,而针对金属/碳微观形貌的微波吸收剂开发研究甚少。这对于采用碳与金属复合微波吸收剂的宽频拓展提出了新的要求。而当前报道的金属/碳基复合微波吸收剂产品较低,环境友好型差,难以被广泛开发和应用。开发新型(产率高、成本低、环境友好)微观结构的金属/碳微波材料能有效拓展微波吸收材料在国防、兵器及民用电子产品中的广泛使用。
技术实现要素:
3.本发明的目的是提供一种银耳状铁基配合物转化电磁波吸收剂的制备方法,其可在较低厚度下实现低频段微波吸收,通过调节不同热解温度形成不同石墨程度的碳金属复合材料,制备得到银耳状铁基配合物转化电磁波吸收剂。
4.本发明采用以下技术方案:一种银耳状铁基配合物转化电磁波吸收剂的制备方法,该制备方法包括如下:
5.步骤s1、制备银耳状铁基配合物fetcnq粉末:
6.分别称取四水合氯化亚铁和7,7,8,8
‑
四氰基对苯二醌二甲烷tcnq,并依次溶于n,n
‑
二甲基甲酰胺dmf中,超声搅拌,形成分散均匀的深黑色混合溶液;在所述混合溶液中,四水合氯化亚铁和7,7,8,8
‑
四氰基对苯二醌二甲烷tcnq的摩尔比为1:1.0~1.1;
7.将所述混合溶液在145~150℃下反应,且混合溶液颜色由深黑色转变为蓝黑色时,将反应好的混合溶液静置12~24h,过滤得沉淀物,清洗所述沉淀物,清洗后在65~70℃下真空干燥,得到蓝黑色银耳状铁基配合物fetcnq粉末;
8.步骤s2、制备电磁波吸收剂fe/c/n纳米复合物:
9.取所述步骤s1中的铁基配合物fetcnq粉末并研磨,最终得到粒径为500~700nm粉
末;
10.将粉末在氩气气氛下热解,具体为:先加热至40℃,再程序升温至800℃,并保温2h热解;再程序降温至400℃,然后自然冷却至室温,得到纳米复合物电磁波吸收剂fe/c/n。
11.进一步地,在步骤s2中,程序升温的速率为2℃/min,程序降温的速率为5℃/min。
12.进一步地,在步骤s2中,所述银耳状铁基配合物fetcnq粉末放置入真空管式炉中热解。
13.进一步地,在步骤s1中,混合溶液在145~150℃下反应9.5~12h;在65~70℃下真空干燥12~24h。
14.本发明的有益效果是:1.直接制备具有微波吸收性能的银耳状纳米片堆积颗粒fetcnq,纳米片在纳米颗粒周围堆积,形成了具有丰富异向界面结构的铁基纳米颗粒可引发更多的电磁波发生散射损耗。2.fe/c/n同轴环的电磁波吸收剂吸波性能显著,其匹配厚度仅为2.9mm,反射损耗为
‑
52.3db,而
‑
10db代表90%电磁波被吸收,有效吸收带宽可达3.1ghz,由14.0
‑
17.1ghz。3.解决了低频下实现在4
‑
5mm处强吸收和宽带吸收的问题,有效吸收频率宽度为3.0ghz。4.原材料廉价,且合成fetcnq配合物产率高,可达40%,有巨大潜在应用价值。
附图说明
15.图1为银耳状铁基配合物fetcnq粉末的表征图;
16.1a为实施例1中制备的银耳状铁基配合物fetcnq粉末的扫描电镜sem图;
17.1b为实施例1中制备的银耳状铁基配合物fetcnq粉末的透射电镜tem图;
18.1c为实施例1中制备的银耳状铁基配合物fetcnq粉末的粉末tg
‑
ms图;
19.1d为实施例1中制备的银耳状铁基配合物fetcnq粉末的粉末x射线衍射pxrd图;
20.图2为实施例1和对比例1中,在不同热解温度下制备得的电磁波吸收剂fe/c/n的pxrd图;
21.图3为实施例1和对比例1制备的fetcnq粉末,以及在不同热解温度下制备得的电磁波吸收剂fe/c/n同轴石蜡基复合材料吸波性能图;
22.图4为对比例2中制备的ni/c/n的吸波性能图;
23.图5为对比例3制备得到的在替换过渡金属下mn/c/n的吸波性能图;
24.图6为电磁波吸收剂fe/c/n在不不同热解温度下的电磁参数。
具体实施方式
25.下面结合附图和具体实施方式对本发明进行详细说明。
26.本发明中所用到的fetcnq粉末为四水合氯化亚铁与7,7,8,8
‑
四氰基对苯二醌二甲烷的配合物,采用本发明中的方法合成得到。采用的四水合氯化亚铁和7,7,8,8
‑
四氰基对苯二醌二甲烷均为试剂级。文中的室温和常温均指的25℃。
27.本发明公开了一种银耳状铁基配合物转化电磁波吸收剂的制备方法,该制备方法包括如下:
28.步骤s1、制备银耳状铁基配合物fetcnq粉末:
29.分别称取四水合氯化亚铁和7,7,8,8
‑
四氰基对苯二醌二甲烷tcnq,并依次溶于n,
n
‑
二甲基甲酰胺dmf中,超声搅拌,形成分散均匀的深黑色混合溶液;在所述混合溶液中,四水合氯化亚铁和7,7,8,8
‑
四氰基对苯二醌二甲烷tcnq的摩尔比为1:1.0~1.1;
30.将所述混合溶液在145~150℃下反应,且混合溶液颜色由深黑色转变为蓝黑色时,将反应好的混合溶液静置12~24h,过滤得沉淀物,清洗所述沉淀物,清洗后在65~70℃下真空干燥,得到蓝黑色银耳状铁基配合物fetcnq粉末;
31.步骤s2、制备电磁波吸收剂fe/c/n纳米复合物:
32.取所述步骤s1中的铁基配合物fetcnq粉末并研磨,最终得到粒径为500~700nm粉末;
33.将粉末在氩气气氛下热解,具体为:银耳状铁基配合物fetcnq粉末放置入真空管式炉中,先加热至40℃,再程序升温至800℃,并保温2h热解;再程序降温至400℃,然后自然冷却至室温,得到纳米复合物电磁波吸收剂fe/c/n。程序升温的速率为2℃/min,程序降温的速率为5℃/min。
34.为验证本发明中的方法,给出如下具体实施例和对比例:
35.实施例1
36.本实施例制备了一种银耳状铁基配合物转化电磁波吸收剂制备方法,其制备方法包括如下步骤:
37.步骤s1、制备银耳状铁基配合物fetcnq粉末:分别称取四水合氯化亚铁和7,7,8,8
‑
四氰基对苯二醌二甲烷tcnq,并依次溶于n,n
‑
二甲基甲酰胺dmf中,超声搅拌,形成分散均匀的深黑色混合溶液;在所述混合溶液中,四水合氯化亚铁和7,7,8,8
‑
四氰基对苯二醌二甲烷tcnq的摩尔比为1:1.0~1.1;在混合溶液中7,7,8,8
‑
四氰基对苯二醌二甲烷tcnq的量要大于等于四水合氯化亚铁的量,是为了保证四水合氯化亚铁反应的更完全。但是,量也不能太多,太多了就造成了浪费。经过多次试验,确定出两者在这样的摩尔比下,四水合氯化亚铁就能完全反应。
38.将所述混合溶液在145~150℃下反应,且混合溶液颜色由深黑色转变为蓝黑色时,将反应好的混合溶液静置12~24h,过滤得沉淀物,清洗所述沉淀物,清洗后在65~70℃下真空干燥,得到蓝黑色银耳状铁基配合物fetcnq粉末。
39.步骤s2、取步骤s1中的铁基配合物fetcnq粉末1.0g,并研磨,最终得到粒径为500~700nm粉末;
40.将银耳状铁基配合物fetcnq粉末放置入真空管式炉中,在氩气气氛下热解,采用程序升温,程序降温的方式进行热解。具体为:先加热至40℃,再程序升温至800℃,并保温2h热解;再程序降温至400℃,然后自然冷却至室温,得到纳米复合物电磁波吸收剂fe/c/n。其中,程序升温的速率为2℃/min,程序降温的速率为5℃/min。
41.对比例1
42.为了验证在不同的热解温度下,制备得到的纳米复合物电磁波吸收剂fe/c/n的性能,将步骤s1中的铁基配合物fetcnq粉末在不同的热解温度下热解,具体为:将银耳状铁基配合物fetcnq粉末放置入真空管式炉中,先加热至40℃,再程序升温至600℃或700℃,并保温2h热解,程序升温的速率为2℃/min,再程序降温至400℃,程序降温的速率为5℃/min,然后自然冷却至室温,得到纳米复合物电磁波吸收剂fe/c/n。
43.将实施例1步骤s1中的制备得到的fetcnq进行测试,如图1中1a所示,为fetcnq的
扫描电镜sem图,由1a中可知fetcnq的微观结构为银耳状。如1b所示,为fetcnq的透射电镜tem图,由1b中可知,fetcnq为纳米片堆积颗粒结构,纳米片在纳米颗粒周围堆积,形成了具有丰富异向界面结构的铁基纳米颗粒可引发更多的电磁波发生散射损耗。如1c所示,为fetcnq粉末的粉末tg
‑
ms图,由图可知,随着温度的升高,fetcnq的热解程度逐渐加深,且在热解温度为800℃时,热解程度最高,热解程度维持恒定,所以选择热解温度为800℃。另外,在热解过程中,失重产量高,在800℃时,失重残留的重量为45%左右。在热解过程中,氮元素以氮气的形式放出。如1d所示,为fetcnq的pxrd粉末衍射图,在2θ角为20~40
°
间产生鼓包,表明fetcnq为晶体结构。
44.对比例2
45.本实施例制备了nitcnq及其衍生物电磁波吸收剂,具体为:将20mmol六水合硝酸镍和20mmol的tcnq分别加入到30ml的dmf溶液中,超声常温下搅拌1h,得浅绿色的配合物。将浅绿色的配合物溶液在150℃下反应9h后,得到米白色粉末,依次用dmf和乙醇洗涤3~5,真空干燥得到nitcnq粉末。
46.将nitcnq粉末1.0g置于陶瓷舟中,随后将装有nitcnq粉末的陶瓷舟转移至管式炉中,设定管式炉的升温速率为2℃/min,气氛为氩气气氛,升温至800℃保温2h,随后设置降温速率为5℃/min,降温至400℃时开始自然降温至室温,即得到粉末吸波剂ni/c/n。
47.对比例3
48.本实施例制备了mntcnq及其衍生物电磁波吸收剂,具体为:将20mmol四水合氯化锰和20mmol的tcnq分别加入到30ml的dmf溶液中,超声常温下搅拌1h,得蓝黑色的混合物。将蓝黑色混合物溶液在150℃下反应9h后,得到黑色粉末,依次用dmf和乙醇洗涤3~5次,真空干燥得到mntcnq粉末。
49.将mntcnq粉末1.0g置于陶瓷舟中,随后将装有mntcnq粉末的陶瓷舟转移至管式炉中,设定管式炉的升温速率为2℃/min,气氛为氩气气氛,升温至800℃保温2h,随后设置降温速率为5℃/min,降温至400℃时,自然降温至室温,即得到粉末吸波剂mn/c/n。
50.将在不同的热解温度下制备得到的fe/c/n粉末及其石蜡基复合相样品分别置于同轴环模具中,其中,石蜡基复合相中fe/c/n粉末和石蜡的质量比为1:1,在同轴环模具中,压缩强度为5~10mpa,保压时间为0.1~0.5h,得到预制的同轴环。同轴环的尺寸均为7.0mm
×
3.04mm。且将样品制备成不同厚度,选择的厚度为1.5mm,2.0mm,2.5mm,3.0mm,3.5mm,4.0mm和4.5mm,对于热解温度800℃下制备的fe/c/n,还制备了厚度为2.9mm的样品,并分别将同组中的两种样品的同轴环分别在矢量网络分析仪下采用同轴线法进行测试。
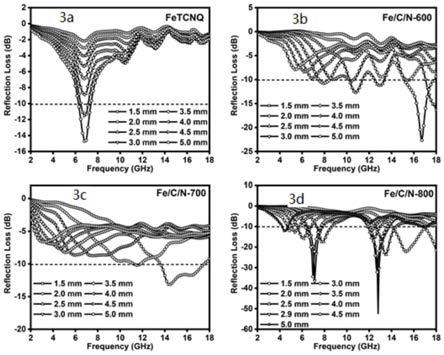
51.对上述样品同轴线法测试结果如下:如图3所示,图中虚线代表有90%的电磁波被吸收,越靠近虚线以下,表明样品对电磁波的吸收性能越好。的由图可知,在800℃的热解温度下制备的fe/c/n的电磁波的吸收性能最好。
52.在图3d中,样品厚度为2.0mm时,fe/c/n的有效吸收带宽eab为3.1ghz,即区间14.0~17.1ghz。而样品厚度为2.9mm时,fe/c/n的反射损耗强度rl值为
‑
52.3db,在则表明在样品厚度为2.9mm处,吸收剂可以吸收99.99%的电磁波,且电磁波的波段为ku波段,表明样品的吸收性能好。
53.本发明中,采用与测试fe/c/n相同的方法测试,将对比例2和3中制备的ni/c/n和mn/c/n分别制备成同轴环,分别在矢量网络分析仪下采用同轴线法进行测试,如图4和5所
示,如图4所示,ni/c/n样品具有较好的电磁波吸收性,在其厚度为2.0mm时,有效吸收带宽eab大于6ghz即区间11.2~18.0ghz。如图5所示,mn/c/n样品在其厚度为2.500mm时,其电磁波吸收强度接近
‑
50db,具有较好的电磁波吸收性。其有效吸收带宽eab小于fe/c/n的有效吸收带宽。
54.在本发明中,是根据如下公式计算得到fe/c/n、ni/c/n和mn/c/n的电磁波吸收性能:
[0055][0056][0057]
以实施例1和对比例1制备的fe/c/n为例,测定不同热解温度下制备的fe/c/n的电磁参数,由图6可知,在x波段下,800℃的热解温度得到的fe/c/n有明显的弛豫现象,其介电实部ε'在波段频率为6
‑
12ghz之间逐渐下降,x波段明显的波动与其微观结构的各项异性密不可分,对应其介电虚部ε"的值在波段频率为6
‑
13ghz间从2.5逐渐增大为14,随后下降为3左右。对比600℃和700℃下热解制备得到的fe/c/n的电磁参数,表现出明显的频散特性,600℃下其介电实部ε'从波段频率为8.5
‑
8左右分布,而介电实部ε'由1
‑
1.5左右变化,从介电虚部ε"得出损耗能力较弱。同样在700℃下热解制备的fe/c/n纳米复合物的介电实部ε'在波段频率为10
‑
19.5变化,表现出强的电存储能力,而其介电虚部ε"的值在19.0
‑
7.5变化,由于介电虚部ε"的值过高表明在高频处,fe/c/n纳米复合物处于屏蔽状态,对电磁波产生了反射,进而电磁吸收能力下降。测定该电磁吸收剂的介电常数实部和介电常数虚部,如图6中a和b所示,如a中所示,介电实部ε'在x波段快速下降,这是因为电磁吸收剂内部由弛豫产生的界面极化造成的。在6b中介电虚部ε"的值快速变化为电子极化造成。如图6中c和d所示,磁导率的实部μ’和虚部μ”呈现明显的波段现象,这是由电磁吸收剂内部异质界面产生的磁滞共振现象影响的,说明电磁波频率的变化速度快于磁畴共振的速度。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。