1.本发明涉及药物化学领域,具体地,涉及一种多取代嘌呤类化合物及其制备方法和应用。
背景技术:
2.作用于rna的腺苷脱氨酶(adenosine deaminases acting on rna enzymes,adars)属于rna编辑酶家族成员之一,它们能作用于rna上特定位点的腺嘌呤核苷酸,发生脱氨作用,将其转化为次黄嘌呤核苷酸,这些次黄嘌呤核苷酸在生物体内会被错误地识别为鸟嘌呤核苷酸而参与转录翻译等一系列生物过程。adars包括三种亚型,adar1,adar2,以及adar3,它们都包含两至三个n端双链rna(double stranded rna,dsrna)结合结构域,以及一个c端保守的脱氨酶催化结构域。adar1与adar2在人体的许多组织器官中都有分布与表达,它们的结构与功能目前被研究的较多。而adar3没有催化脱氨活性,主要表达于中枢神经系统中。
3.人adar1蛋白由位于1号染色体q21区域的adar1基因表达,adar1基因由两种启动子驱动转录,一种在基因组上组成性的表达,产生p110蛋白,另一种是干扰素诱导的启动子,促进转录产生p150蛋白,因此p150蛋白是干扰素诱导型的。相比于p110蛋白,p150蛋白多了一段z
‑
dna结合结构域zα,并且只有zα结构域能结合z
‑
dna/rna。
4.adar1介导的a
‑
to
‑
i编辑事件广泛发生于各组织器官中的细胞中,由于次黄嘌呤核苷酸在生物过程中被错误地识别为鸟苷酸,发生在mrna编码区的a
‑
to
‑
i编辑事件可能会导致蛋白氨基酸的改变,产生结构和功能突变的蛋白。但大部分a
‑
to
‑
i编辑事件发生在rna的内含子和3
′
非翻译区(3
′‑
utrs),影响着mrna的剪切,降解以及翻译调控,目前adar1介导的发生在非编码区的编辑事件的生物意义还远未研究清楚。
5.adar1的表达和活性在许多癌症中有显著的上调,这些由adar1介导的a
‑
to
‑
i编辑事件在正常组织和癌症组织中有明显的差异,并且与癌症病人的临床预后呈显著的负相关。在许多癌症的细胞系中,如肺癌,肝癌,乳腺癌,多发性骨髓瘤,胃癌,胰腺癌等,通过短发卡rna(short
‑
hairpin rna)敲低adar1,利用crisper
‑
cas9技术敲除adar1,降低adar1蛋白的表达,发现细胞的增殖活性,存活,转移,侵袭能力都显著下降。这主要是由于未被编辑过的dsrna会错误地引起细胞的抗病毒反应,被细胞内的mda5识别,引起下游mavs激活分泌干扰素,导致免疫反应,被pkr识别,引起翻译抑制,导致细胞死亡,而在许多癌症中adar1蛋白水平和活性过度升高,dsrna被编辑后不会被mda5和pkr识别导致细胞死亡和免疫反应,从而使癌症细胞存活和过度增殖。此外,在黑色素瘤中敲除adar1会增强肿瘤对放疗和免疫抑制剂如pd
‑
1单克隆抗体的敏感性,并且能克服对免疫疗法的耐药。这揭示adar1是一个很有希望的癌症靶标。但目前还未报道直接作用于adar1的小分子抑制剂。因此,发现第一个adar1小分子抑制剂是很有治疗意义的。
技术实现要素:
6.发明目的:本发明的目的是提供了一种多取代嘌呤类化合物及其制备方法;本发明还提供该类化合物的应用。
7.技术方案:本发明所述的一种多取代嘌呤类化合物,如通式(i)所示的化合物或其药学上可接受的盐;
[0008][0009]
式中,r1选自氢、c1‑
c3烷基;
[0010]
r2选自卤素、
‑
sch2ch2cooh;
[0011]
r3选自氢、卤素、c1‑
c3烷基;
[0012]
r4选自c1‑
c3烷基、
‑
ch2p(o)(och2ch3)2、
[0013]
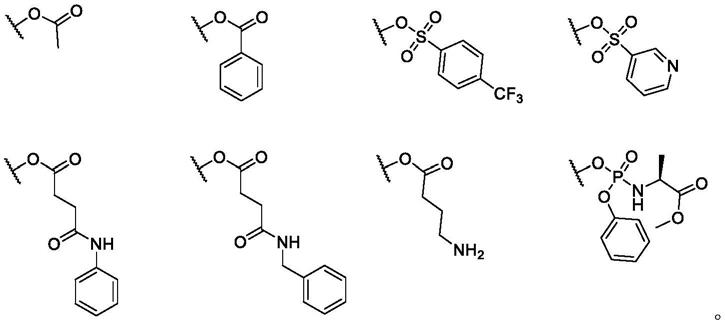
r5选自以下基团:
[0014][0015]
r6选自于羟基、卤素、
‑
sch2ch2cooh或以下基团:
[0016][0017]
进一步的,
[0018]
r1选自氢、甲基;
[0019]
r2选自氟、
‑
sch2ch2cooh;
[0020]
r3选自氢、氯、甲基;
[0021]
r4选自c1‑
c3烷基、
‑
ch2p(o)(och2ch3)2、
[0022]
r5选自以下基团:
[0023][0024]
r6选自于羟基、卤素、
‑
sch2ch2cooh或以下基团:
[0025][0026]
进一步的,
[0027]
r1选自氢、甲基;
[0028]
r2选自氟、
‑
sch2ch2cooh;
[0029]
r3选自氢、氯、甲基;
[0030]
r4选自甲基、丙基、
‑
ch2p(o)(och2ch3)2、
[0031]
r5选自以下基团:
[0032][0033]
r6选自于羟基、卤素、
‑
sch2ch2cooh或以下基团:
[0034][0035]
进一步的,
[0036]
r1选自氢;
[0037]
r2选自氟;
[0038]
r3选自氢;
[0039]
r4选自甲基、丙基、
‑
ch2p(o)(och2ch3)2、
[0040]
r5选自以下基团:
[0041][0042]
r6选自于羟基、卤素、
‑
sch2ch2cooh或以下基团:
[0043][0044]
进一步的,
[0045]
r1选自氢;
[0046]
r2选自氟;
[0047]
r3选自氢;
[0048]
r4选自甲基或
[0049]
r6选自于羟基、卤素、
‑
sch2ch2cooh或以下基团:
[0050][0051]
进一步的,
[0052]
所述化合物选自i
‑
1至i
‑
21:
[0053]
[0054]
[0055][0056]
进一步的,所述药学上可接受的盐为通式(i)化合物的酸加成盐,其中用于成盐的酸包括无机酸及有机酸,所述无机酸包括盐酸、硫酸、磷酸和甲磺酸,有机酸包括乙酸、三氯乙酸、丙酸、丁酸、马来酸、对甲苯磺酸、苹果酸、丙二酸、肉桂酸、柠檬酸、富马酸、樟脑酸、二葡糖酸、天冬氨酸和酒石酸。
[0057]
进一步的,所述的药学上可接受的盐为盐酸盐。
[0058]
进一步的,一种多取代嘌呤类化合物的制备方法。
[0059]
进一步的,一种药用组合物,包含通式(i)化合物或其药学上可接受的盐或其异构
体,以及药学上可接受的载体。
[0060]
药学上可接受的载体指的是对有机体不引起明显的刺激性和不干扰所给予化合物的生物活性和性质的赋形剂或稀释剂。
[0061]
进一步的,一种多取代嘌呤类化合物在制备用于预防和/或治疗癌症或肿瘤相关疾病的药物中的应用,所述癌症或肿瘤相关疾病包括前列腺癌、白血病、乳腺癌、多发性骨髓瘤、肺癌、胃癌、卵巢癌、结肠癌、肝癌、胰腺癌以及人神经胶质瘤。
[0062]
本发明所述的通式(i)化合物或其药学上可接受的盐,具有adar1靶点抑制活性,对肿瘤具有治疗效果。
[0063]
本发明中的术语除特别说明外,一般具有如下的含义。
[0064]
术语“烷基”表示具有所述数目碳原子的直链或支链饱和烃基。
[0065]
术语“c1‑
c3烷基”是指具有1
‑
3个碳原子的直链或支链饱和烃基;c1‑
c3烷基包括但不限于甲基、乙基、正丙基、异丙基。
[0066]“p(o)”表示
“‑
p(o)
‑”
,具体为磷氧双键。
[0067]
术语“卤素”为氟、氯、溴或碘;优选为氟、氯、溴。
[0068]
本发明还公开了通式(ⅰ)化合物的制备方法。
[0069]
有益效果:本发明与现有技术相比,本发明公开了一种新的通式(ⅰ)所示的化合物,此类化合物具有抑制adar1活性,为靶向adar1疗法治疗癌症提供了可成药的化合物;此类化合物能够同时抑制癌症增殖和转移,治疗效果好、毒性低、不易产生耐药性问题,可用于治疗癌症或肿瘤相关疾病;本发明还公开了通式(ⅰ)化合物的制备方法。
附图说明
[0070]
图1是本发明化合物与adar1蛋白结合常数结果图;
[0071]
图2是本发明化合物对adar1脱氨酶活性抑制能力的结果图;
[0072]
图3是本发明急性毒性测定中小鼠的体重变化示意图;
[0073]
图4是本发明急性毒性测定中he染色结果图;
[0074]
图5是本发明对前列腺癌肿瘤体积的结果图。
具体实施方式
[0075]
下面结合附图对本发明作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
[0076]
实施例1:(2r,3r,4s,5s)
‑2‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑5‑
(氯甲基)四氢呋喃
‑
3,4
‑
二醇(i
‑
1)的合成:
[0077][0078]
在0℃下,2
‑
氟腺苷a
‑
1(5.70g,20mmol)溶于乙腈(80ml),加入吡啶(3.22ml,
40mmol),再向悬浮液中缓慢滴加二氯亚砜(7.25ml,100mmol),搅拌4h后提至室温,过夜反应,tlc监测原料消耗完全,减压旋蒸除去溶剂,重新加入甲醇(120ml)、水(12ml)、氨水(24ml),搅拌0.5h,减压旋蒸浓缩,水相析出产物,过滤得滤饼,60℃下重新溶于少量甲醇,滴加二氯甲烷,冷却析出固体抽滤得滤饼,冷甲醇洗涤得产物i
‑
1白色固体(5.45g,90%)。1h nmr(400mhz,dmso
‑
d6)δ8.34(s,1h),7.92(s,2h),5.84(d,j=5.6hz,1h),5.58(s,2h),4.67(t,j=5.4hz,1h),4.26
–
4.16(m,1h),4.10(q,j=5.4,5.0hz,1h),3.99
–
3.81(m,2h)。
[0079]
实施例2:9
‑
((3ar,4r,6r,6ar)
‑6‑
(((叔丁基二甲基甲硅烷基)氧基)甲基)
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3]二恶唑
‑4‑
基)
‑2‑
氟
‑
9h
‑
嘌呤
‑6‑
胺(i
‑
2)的合成:
[0080][0081]
步骤一,2
‑
氟腺苷(1.48g,5mmol)溶于无水丙酮(200ml)形成悬浮液,加入无水对甲苯磺酸(4.31g,25mmol)形成澄清溶液,向此溶液中加入二甲氧基丙烷(1.04g,10mmol),将混合物在氮气氛围下室温搅拌4h后,将冷的饱和nahco3溶液(100ml)加入上述混合物中;减压除去挥发物,并将残余物干燥;将得到的固体溶解在丙酮(400ml)中,搅拌1h,然后过滤;除去挥发物,并将粗产物通过柱色谱法纯化获得白色固体a2(1.50g,95%);1h nmr(400mhz,dmso
‑
d6)δ8.33(s,1h),7.92(s,2h),6.03(d,j=2.9hz,1h),5.29(dd,j=6.2,2.9hz,1h),5.10(s,1h),4.93(dd,j=6.2,2.6hz,1h),4.27
–
4.10(m,1h),3.63
–
3.44(m,2h),1.54(s,3h),1.33(s,3h);
[0082]
步骤二,0℃,n2保护下,a2(0.66g,2mmol)溶于10ml dmf中,加入咪唑(0.41g,6mmol)和叔丁基二甲基氯硅烷(0.45g,3mmol),反应2h后,提至室温,过夜反应;tlc监测原料消耗完全,用甲醇(2.0ml)处理,搅拌30分钟后,减压除去溶剂;残留物经硅胶柱层析纯化,得到白色产物i
‑
2(0.84g,95%)。1h nmr(400mhz,chloroform
‑
d)δ8.02(s,1h),6.41(s,2h),6.10(d,j=2.7hz,1h),5.20(dd,j=6.1,2.7hz,1h),4.95(dd,j=6.2,2.6hz,1h),4.40(q,j=3.9hz,1h),3.99
–
3.70(m,2h),1.63(s,3h),1.40(s,3h),0.87(s,9h),0.04(s,6h)。
[0083]
实施例3:((3ar,4r,6r,6ar)
‑6‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3]二恶唑
‑4‑
基)乙酸甲酯(i
‑
3)的合成:
[0084]
a2(0.66g,2mmol)溶于10ml dmf中,加入三乙胺(0.83ml,6mmol)、乙酸酐(0.21ml,2.2mmol)和dmap(0.05g,0.4mmol),室温过夜反应。tlc监测原料消耗完全,饱和nh4cl水溶液(5ml)淬灭反应,将混合物用乙酸乙酯(3
×
30ml)萃取,合并有机层用饱和nacl水溶液洗涤,无水na2so4干燥,过滤并减压浓缩,硅胶柱色谱纯化,得到化合物i
‑
3(0.66g,90%)。1h nmr(400mhz,dmso
‑
d6)δ8.28(s,1h),7.92(d,j=26.2hz,2h),6.11(d,j=2.4hz,1h),5.41(dd,j=6.2,2.5hz,1h),5.00(dd,j=6.2,3.3hz,1h),4.45
–
4.30(m,1h),4.30
–
4.08(m,2h),1.96(s,3h),1.54(s,3h),1.34(s,3h)。
[0085]
实施例4:((3ar,4r,6r,6ar)
‑6‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3]二恶唑
‑4‑
基)苯甲酸甲酯(i
‑
4)的合成:
[0086]
方法同i
‑
3的合成,收率92%。1h nmr(400mhz,dmso
‑
d6)δ8.26(s,1h),7.87(dd,j=8.3,1.4hz,4h),7.69
–
7.59(m,1h),7.48(t,j=7.8hz,2h),6.14(d,j=2.4hz,1h),5.47(dd,j=6.3,2.4hz,1h),5.14(dd,j=6.3,3.0hz,1h),4.59
–
4.47(m,2h),4.48
–
4.38(m,1h),1.56(s,3h),1.36(s,3h)。
[0087]
实施例5:((3ar,4r,6r,6ar)
‑6‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3]二恶唑
‑4‑
基)甲基4
‑
(三氟甲基)苯磺酸酯(i
‑
5)的合成:
[0088]
方法同i
‑
3的合成,收率89%。1h nmr(400mhz,dmso
‑
d6)δ8.23(s,1h),8.06
–
7.83(m,6h),6.10(d,j=2.3hz,1h),5.30(dd,j=6.3,2.3hz,1h),4.93(dd,j=6.3,3.3hz,1h),4.47
–
4.31(m,3h),1.51(s,3h),1.29(s,3h)。
[0089]
实施例6:((2r,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)乙酸甲酯(i
‑
6)的合成:
[0090][0091]
取化合物i
‑
3(0.37g,1mmol)加入到10ml的单口瓶中,冰浴条件下滴加5ml三氟乙酸:水=4:1,提至室温过夜反应;tlc监测原料消耗完全,饱和nahco3水溶液中和反应体系,减压旋蒸除去溶剂,硅胶柱色谱纯化,得到产物i
‑
6(0.31g,96%)。1h nmr(400mhz,methanol
‑
d4)δ8.18(s,1h),5.91(d,j=4.5hz,1h),4.72(t,j=4.9hz,1h),4.45
–
4.27(m,3h),4.22(td,j=5.1,3.6hz,1h),2.06(s,3h)。
[0092]
实施例7:((2r,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)苯甲酸甲酯的合成:
[0093]
方法同i
‑
6的合成,收率94%。1h nmr(300mhz,dmso
‑
d6)δ8.29(s,1h),8.05
–
7.76(m,4h),7.72
–
7.63(m,1h),7.52(t,j=7.7hz,2h),5.86(d,j=4.7hz,1h),5.78
–
5.22(m,2h),4.68(t,j=5.0hz,1h),4.59(dd,j=12.0,3.7hz,1h),4.47(dd,j=12.0,5.5hz,1h),4.41(t,j=5.1hz,1h),4.27
–
4.19(m,1h)。
[0094]
实施例8:((2r,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)4
‑
(三氟甲基)苯磺酸甲酯(i
‑
8)的合成:
[0095]
方法同i
‑
6的合成,收率93%。1h nmr(400mhz,dmso
‑
d6)δ8.24(s,1h),8.06(d,j=8.2hz,2h),7.92(d,j=8.2hz,4h),5.76(d,j=5.1hz,1h),5.72
–
5.33(m,2h),4.55(t,j=5.1hz,1h),4.48
–
4.40(m,2h),4.18(t,j=4.7hz,1h),4.12
–
4.06(m,1h)。
[0096]
实施例9:((2r,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)吡啶
‑3‑
磺酸甲酯(i
‑
9)的合成:
[0097]
方法同i
‑
6的合成,收率96%。1h nmr(300mhz,dmso
‑
d6)δ8.99(dd,j=2.5,0.8hz,1h),8.83(dd,j=4.8,1.6hz,1h),8.25(ddd,j=8.1,2.5,1.6hz,1h),8.20(s,1h),7.90(s,2h),7.58(ddd,j=8.1,4.9,0.9hz,1h),5.76(d,j=5.2hz,1h),5.62(d,j=5.8hz,1h),5.46(d,j=5.3hz,1h),4.54(q,j=5.3hz,1h),4.42(qd,j=11.0,5.0hz,2h),4.17(q,j=4.9hz,1h),4.11
–
4.05(m,1h)。
[0098]
实施例10:((2r,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)甲基4
‑
氧代
‑4‑
(苯基氨基)丁酸(i
‑
10)的合成:
[0099][0100]
步骤一,取丁二酸酐(0.50g,5mmol)与苯胺(0.47g,5mmol)溶于甲苯中,加热至110℃回流2h,冷却抽滤,乙醚洗涤,收取滤饼干燥得产物b1(0.92g,95%);1h nmr(300mhz,dmso
‑
d6)(300mhz,)δ12.13,9.95,7.58(d,j=8.0hz),7.28(t,j=7.8hz),7.01(t,j=7.4hz),2.62
–
2.52(m);
[0101]
步骤二,a2(0.66g,2mmol)溶于10ml dmf中,分别加入b1(0.58g,3mmol)、二环己基碳二亚胺(0.83g,4mmol)、4
‑
二甲氨基吡啶(48mg,0.4mmol),室温过夜反应;tlc监测原料消耗完全,用饱和nh4cl水溶液(5ml)淬灭;将混合物用二氯甲烷(3
×
30ml)萃取,合并有机层用饱和nacl水溶液洗涤,无水na2so4干燥,过滤并减压浓缩,硅胶柱色谱纯化,得到化合物b2(0.90g,90%);1h nmr(400mhz,dmso
‑
d6)δ9.98(s,1h),8.31(s,1h),7.93(d,j=28.4hz,2h),7.56(d,j=8.0hz,2h),7.27(t,j=7.7hz,2h),7.01(t,j=7.3hz,1h),6.09(d,j=2.6hz,1h),5.38(dd,j=6.2,2.6hz,1h),4.99(dd,j=6.2,3.3hz,1h),4.37(q,j=3.9hz,1h),4.31
–
4.12(m,2h),2.59(s,4h),1.53(s,3h),1.31(s,3h);
[0102]
步骤三,取化合物b2(0.50g,1mmol)加入到10ml的单口瓶中,冰浴条件下滴加5ml三氟乙酸:水=4:1,提至室温过夜反应;tlc监测原料消耗完全,饱和nahco3水溶液中和反应体系,减压旋蒸除去溶剂,硅胶柱色谱纯化,得到产物i
‑
10(0.43g,93%)。1h nmr(400mhz,dmso
‑
d6)δ9.98(s,1h),8.33(s,1h),7.89(d,j=29.7hz,2h),7.56(d,j=7.6hz,2h),7.28(t,j=7.9hz,2h),7.01(t,j=7.4hz,1h),5.81(d,j=5.0hz,1h),5.61(s,1h),5.41(s,1h),4.57(t,j=5.1hz,1h),4.34(dd,j=11.9,3.7hz,1h),4.26
–
4.15(m,2h),4.14
–
4.05(m,1h),2.62(t,j=3.9hz,4h)。
[0103]
实施例11:((2r,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)甲基4
‑
(苄基氨基)
‑4‑
氧代丁酸酯(i
‑
11)的合成:
[0104]
方法同i
‑
10的合成,收率94%。1h nmr(400mhz,methanol
‑
d4)δ8.21(s,1h),7.35
–
7.14(m,5h),5.90(d,j=4.6hz,1h),4.70(t,j=4.9hz,1h),4.44
–
4.33(m,5h),4.23(q,j=4.8hz,1h),2.72
–
2.64(m,2h),2.59
–
2.52(m,2h)。
[0105]
实施例12:((2r,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)4
‑
氨基丁酸甲酯(i
‑
12)的合成:
[0106]
方法同i
‑
10的合成,收率90%。1h nmr(400mhz,methanol
‑
d4)δ8.18(s,1h),5.90(d,j=4.4hz,1h),4.77
–
4.72(m,1h),4.49
–
4.32(m,3h),4.27
–
4.17(m,1h),3.00
–
2.92(m,2h),2.51(t,j=7.1hz,2h),1.92(p,j=7.2hz,2h)。
[0107]
实施例13:(((5
‑
(6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)甲氧基)(苯氧基)磷酰基)
‑
l
‑
丙氨酸甲酯(i
‑
13)的合成:
[0108][0109]
步骤一,
‑
78℃,n2保护下,c1(2.95g,14mmol)与c2(1.95g,14mmol)溶于无水二氯甲烷(40ml)中,缓慢滴加无水三乙胺(2.83g,28mmol),缓慢升至室温,继续反应24h,tlc监测原料消耗完全,减压旋蒸除去溶剂,重新溶于40ml无水乙醚并过滤,收集滤液,旋蒸浓缩得粗油c3,无需纯化直接使用;
[0110]
步骤二,取a2(0.65g,2mmol)与n
‑
甲基咪唑(0.49g,6mmol)溶于20ml thf中,于
‑
78℃下滴加c3(0.89g,3.2mmol)的thf(5ml)溶液,搅拌2h,缓慢升至室温继续反应24h,用甲醇淬灭,减压旋蒸除去溶剂,硅胶柱色谱纯化,得到一对非对映异构体c4(0.93g,82%);1h nmr(400mhz,chloroform
‑
d)δ7.94(d,j=4.2hz,1h),7.32
–
7.23(m,2h),7.20
–
7.08(m,3h),6.56(s,2h),6.04(dd,j=9.1,2.7hz,1h),5.28(dd,j=6.3,2.5hz,0.5h),5.09(dd,j=6.3,2.8hz,0.5h),5.04(dd,j=6.3,3.3hz,0.5h),4.94(dd,j=6.3,2.9hz,0.5h),4.55
–
4.43(m,1h),4.40
–
4.27(m,2h),4.14
–
3.93(m,1h),3.67(d,j=1.2hz,3h),1.60(d,j=4.4hz,3h),1.36(d,j=13.4hz,3h),1.33
–
1.28(m,3h);
[0111]
步骤三,取化合物c4(0.57g,1mmol)加入到10ml的单口瓶中,冰浴条件下滴加5ml三氟乙酸:水=4:1,提至室温过夜反应;tlc监测原料消耗完全,饱和nahco3水溶液中和反应体系,减压旋蒸除去溶剂,硅胶柱色谱纯化,得到产物,一对非对映异构体i
‑
13(0.47g,90%)。1h nmr(400mhz,methanol
‑
d4)δ8.18(d,j=13.7hz,1h),7.44
–
6.92(m,5h),5.93(dd,j=4.8,3.2hz,1h),4.63(dt,j=15.8,5.0hz,1h),4.48
–
4.18(m,4h),4.03
–
3.78(m,1h),3.64(d,j=2.0hz,3h),1.32
–
1.29(m,1.5h),1.23(dd,j=7.2,1.3hz,1.5h)。
[0112]
实施例14:(2r,3r,4s,5r)
‑2‑
(2
‑
氟
‑6‑
(甲基氨基)
‑
9h
‑
嘌呤
‑9‑
基)
‑5‑
(甲氧基甲基)四氢呋喃
‑
3,4
‑
二醇(i
‑
14)的合成:
[0113][0114]
步骤一,0℃下,将a2(0.65g,2mmol)的无水dmf(10ml)溶液滴加至取氢化钠
(0.16g,4mmol)的无水dmf(10ml)溶液中,升至室温反应2h后,滴加碘甲烷(0.57g,4mmol)室温过夜反应,用饱和nh4cl水溶液(5ml)淬灭;将混合物用二氯甲烷(3
×
30ml)萃取,合并有机层用饱和nacl水溶液洗涤,无水na2so4干燥,过滤并减压浓缩,硅胶柱色谱纯化,得到化合物d1(0.52g,73%);1h nmr(400mhz,chloroform
‑
d)δ7.70(s,1h),5.78(d,j=4.9hz,1h),5.65(dd,j=11.6,2.0hz,1h),5.23
–
5.16(m,1h),5.12(dd,j=6.0,1.4hz,1h),4.50(d,j=1.7hz,1h),3.98(dt,j=12.8,1.8hz,1h),3.92
–
3.61(m,4h),3.27(s,3h),1.64(s,3h),1.38(s,3h);
[0115]
步骤二,取化合物c4(0.57g,1mmol)加入到10ml的单口瓶中,冰浴条件下滴加5ml三氟乙酸:水=4:1,提至室温过夜反应。tlc监测原料消耗完全,饱和nahco3水溶液中和反应体系,减压旋蒸除去溶剂,硅胶柱色谱纯化,得到产物i
‑
14(0.29g,91%)。1h nmr(400mhz,dmso
‑
d6)δ8.38(s,1h),5.81(d,j=5.7hz,1h),4.49(t,j=5.3hz,1h),4.15
–
4.11(m,1h),3.94(q,j=3.9hz,1h),3.78
–
3.61(m,4h),3.55(dd,j=12.0,3.9hz,1h),3.17(s,3h)。
[0116]
实施例15:3
‑
((((2s,3s,4r,5r)
‑5‑
(6
‑
氨基
‑2‑
((2
‑
羧乙基)硫代)
‑
9h
‑
嘌呤
‑9‑
基)
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)甲基)硫代)丙酸(i
‑
15)的合成:
[0117][0118]
n2氛围下,3
‑
巯基丙酸(1.11g,10.5mmol)溶于1m naoh水溶液(18ml),加入i
‑
1(0.91g,3mmol),加热至105℃,过夜回流反应,tlc监测原料消耗完全,乙酸调节ph至5~6,固体析出,抽滤,水和冷甲醇洗涤,干燥,得产物白色固体(1.14g,83%)。1h nmr(400mhz,dmso
‑
d6)δ8.24(s,1h),7.41(s,2h),5.81(d,j=5.9hz,1h),4.76(t,j=5.5hz,1h),4.21
–
4.06(m,1h),4.04
–
3.96(m,1h),3.32
–
3.13(m,2h),2.95
–
2.79(m,2h),2.69(t,j=6.4hz,4h),2.50
–
2.41(m,2h)。
[0119]
实施例16:(2r,3r,4s,5r)
‑2‑
(6
‑
氨基
‑2‑
氟
‑8‑
甲基
‑
9h
‑
嘌呤
‑9‑
基)
‑5‑
(羟甲基)四氢呋喃
‑
3,4
‑
二醇(i
‑
16)的合成:
[0120][0121]
步骤一,取a1(0.57g,2mmol)与咪唑(0.82g,12mmol)溶于无水dmf(20ml)中,添加tbscl(1.2g,8mmol),升至50℃过夜反应,tlc监测原料消耗完全,用饱和nh4cl水溶液(5ml)淬灭;将混合物用二氯甲烷(3
×
30ml)萃取,合并有机层用饱和nacl水溶液洗涤,无水na2so4干燥,过滤并减压浓缩,硅胶柱色谱纯化,得到化合物i
‑
12(1.17g,93%);1h nmr(400mhz,chloroform
‑
d)δ8.12(s,1h),6.14(s,2h),5.93(d,j=5.1hz,1h),4.63(t,j=4.7hz,1h),
4.31(t,j=4.0hz,1h),4.13(q,j=3.7hz,1h),4.03(dd,j=11.3,4.3hz,1h),3.78(dd,j=11.4,2.9hz,1h),0.94(d,j=9.2hz,18h),0.81(s,9h),0.12(dd,j=13.6,2.9hz,12h),
‑
0.02(s,3h),
‑
0.20(s,3h);
[0122]
步骤二,
‑
78℃下,化合物e1(0.94g,1.5mmol)溶于无水thf(10ml)中,缓慢滴加二异丙基氨基锂(7.5mmol),加毕搅拌3h后,再次滴加碘甲烷(0.64g,4.5mmol),反应2h,tlc监测原料消耗完全,用饱和nh4cl水溶液(5ml)淬灭;将混合物二氯甲烷(3
×
25ml)萃取,合并有机层用饱和nacl水溶液洗涤,无水na2so4干燥,过滤并减压浓缩,硅胶柱色谱纯化,得到化合物e2(0.72g,75%);1hnmr(300mhz,chloroform
‑
d)δ5.93(s,2h),5.72(d,j=6.1hz,1h),5.30(dd,j=6.1,4.4hz,1h),4.47(dd,j=4.4,2.2hz,1h),4.07(dd,j=4.4,2.2hz,2h),3.78
–
3.67(m,1h),2.58(s,3h),0.95(s,9h),0.85(s,9h),0.78(s,9h),0.15(d,j=1.1hz,6h),0.06
–‑
0.01(m,6h),
‑
0.05(s,3h),
‑
0.35(s,3h);
[0123]
步骤三,化合物e2(0.64g,1mmol)溶于无水thf(10ml)中,于0℃下滴加四丁基氟化铵(4mmol)的thf溶液,升至室温反应3h,tlc监测原料消耗完全,减压旋蒸除去溶剂,用二氯甲烷(3
×
15ml)萃取,合并有机层用饱和nacl水溶液洗涤,无水na2so4干燥,过滤并减压浓缩,硅胶柱色谱纯化,得到化合物i
‑
16(0.27g,90%)。1h nmr(400mhz,methanol
‑
d4)δ5.85(d,j=7.1hz,1h),4.88(s,1h),4.32(dd,j=5.3,2.0hz,1h),4.14(q,j=2.7hz,1h),3.87(dd,j=12.6,2.7hz,1h),3.72(dd,j=12.5,3.1hz,1h),2.60(s,3h)。
[0124]
实施例17:(2r,3r,4s,5r)
‑2‑
(6
‑
氨基
‑8‑
氯
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑5‑
(羟甲基)四氢呋喃
‑
3,4
‑
二醇(i
‑
17)的合成:
[0125][0126]
取a(0.30g,1mmol)溶于5ml dmf中,加入n
‑
氯代丁二酰亚胺(0.19g,1.4mmol),升温至55℃反应3h,tlc监测原料消耗完全,二氯甲烷(3
×
15ml)萃取,合并有机层用饱和nacl水溶液洗涤,无水na2so4干燥,过滤并减压浓缩,硅胶柱色谱纯化,得到化合物i
‑
17(0.30g,95%)。1h nmr(300mhz,dmso
‑
d6)δ8.09(d,j=29.7hz,2h),5.79(d,j=6.4hz,1h),5.02
–
4.95(m,1h),4.19(dd,j=5.2,2.9hz,1h),3.93(q,j=4.9hz,1h),3.58(ddd,j=44.2,11.9,5.1hz,2h)。
[0127]
实施例18:(2r,3r,4s,5s)
‑2‑
(6
‑
氨基
‑8‑
氯
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)
‑5‑
(氯甲基)四氢呋喃
‑
3,4
‑
二醇(i
‑
18)的合成:
[0128]
方法同i
‑
17的合成,收率94%。1h nmr(300mhz,dmso
‑
d6)δ8.08(d,j=30.2hz,2h),5.83(d,j=5.5hz,1h),5.66(d,j=5.8hz,1h),5.55(d,j=5.3hz,1h),5.04(q,j=5.4hz,1h),4.33(q,j=4.7hz,1h),4.14
–
4.02(m,1h),3.99
–
3.75(m,2h)。
[0129]
实施例19:2
‑
氟
‑9‑
甲基
‑
9h
‑
嘌呤
‑6‑
胺(i
‑
19)的合成:
[0130][0131]
取2
‑
氟腺嘌呤(0.15g,1mmol)溶于dmf(5ml)中,加入碳酸铯(0.39g,1.2mmol)和碘甲烷(0.17g,1.2mmol);将反应混合物加热至60℃持续24h,并用水(10ml)淬灭;过滤沉淀物并真空干燥,得到产物白色粉末i
‑
19(0.15g,91%)。1h nmr(400mhz,dmso
‑
d6)δ8.08(s,1h),7.77(s,2h),3.68(s,3h)。
[0132]
实施例20:2
‑
氟
‑9‑
丙基
‑
9h
‑
嘌呤
‑6‑
胺(i
‑
20)的合成:
[0133]
方法同i
‑
19的合成,收率93%。1h nmr(300mhz,dmso
‑
d6)δ8.14(s,1h),7.76(s,2h),4.04(t,j=7.1hz,2h),1.79(h,j=7.3hz,2h),0.84(t,j=7.4hz,3h)。
[0134]
实施例21:((6
‑
氨基
‑2‑
氟
‑
9h
‑
嘌呤
‑9‑
基)甲基)膦酸二乙酯(i
‑
21)的合成:
[0135][0136]
方法同i
‑
19的合成,收率81%。1h nmr(400mhz,methanol
‑
d4)δ8.05(d,j=0.8hz,1h),4.70(d,j=11.9hz,2h),4.26
–
4.09(m,4h),1.27(t,j=7.1hz,6h)。
[0137]
三、生物学评价实验:
[0138]
(1)、癌细胞体外增殖抑制作用测定:
[0139]
化合物对三阴性乳腺癌mda
‑
mb
‑
231细胞系、多发性骨髓瘤rpmi8226细胞系、急性髓系白血病(hl60,k562,nb4)细胞系、胃癌细胞系hgc
‑
27、肝癌hepg2细胞系、人神经胶质瘤(u251)细胞系、非小细胞肺癌a549细胞系和前列腺癌(pc
‑
3、du
‑
145、lncap、vcap、22rv1)细胞系,人胚肾细胞hek293,人正常前列腺上皮细胞rwpe1等16种细胞增殖抑制作用通过以下的方法进行测试。
[0140]
实验步骤:
[0141]
按照cck
‑
8法进行测定化合物对多种癌细胞增殖的抑制作用,并得出化合物抑制细胞增殖活性的半数抑制浓度ic
50
值;
[0142]
1)、将对数生长期细胞以5000
‑
20000cells/孔接种于96孔板,置于37℃,5%co2条件下培养12
‑
24小时;
[0143]
2)、向培养板加入梯度稀释的不同浓度的待测化合物溶液100μl,将培养板在37℃,5%co2培养箱条件下孵育72小时;
[0144]
3)、孵育结束前4h,每孔加入10μl cck
‑
8溶液(5mg/ml)。孵育结束后,用酶标仪测定od
450
,抑制率=(对照组od值-实验组od值)/对照组od值
×
100%;
[0145]
4)、得出数据后,graphpad prism 8.0拟合得出ic
50
。
[0146]
从实验结果可以看出,本发明实例化合物对前列腺癌du
‑
145细胞系具有很强的抑制作用,具体结果见表1;此外,实施例1的化合物(i
‑
1)还对其它多种癌症细胞具有很强的抑制作用,测得的ic
50
值见表2;可以看出化合物i
‑
1对三阴性乳腺癌mda
‑
mb
‑
231细胞系、多发性骨髓瘤rpmi8226细胞系、急性髓系白血病(hl60,k562,nb4)细胞系、胃癌细胞系hgc
‑
27、肝癌hepg2细胞系、人神经胶质瘤(u251)细胞系、非小细胞肺癌a549细胞系和前列腺癌
(pc
‑
3、du
‑
145、lncap、vcap、22rv1)细胞系有强烈的增殖抑制效果,对正常细胞rwpe1和hek293细胞只有较弱增殖抑制作用。
[0147]
表1化合物对前列腺癌du
‑
145细胞增殖活性抑制作用
[0148][0149][0150]
表2化合物(i
‑
1)对多种癌症细胞增殖活性抑制ic
50
[0151][0152]
(2)、adar1蛋白结合分析(mst实验):
[0153]
通过mst微量热泳动仪(monolith nt.115)来进行化合物与adar1亲和力测定;经三次重复试验计算出实施例1化合物化合物i
‑
1与adar1蛋白的结合常数kd为7.24
±
2.88μm(见图1),显示本发明实例化合物与adar1蛋白有较好的结合。
[0154]
(3)、adar1脱氨酶活性检测方法:
[0155]
本实验采用abcam公司的adenosine deaminase(ada)activity assay kit(cat.ab204695)进行检测;取线性增长区间两个时间点的荧光值差即可计算待测实施例1化合物i
‑
1对adar1的脱氨酶活性的抑制能力ic
50
为0.87μm(见图2),显示本发明实例化合物对adar1蛋白脱氨活性活性具有较强的抑制作用。
[0156]
(4)、化合物急性毒性测定:
[0157]
受试动物:icr小鼠;18
‑
22g;雌性;共70只;
[0158]
组别剂量设置:预实验显示受试药物有一定毒性,药物腹腔注射500mg/kg剂量可引起4/4只小鼠死亡,而在腹腔注射100mg/kg剂量下引起0/4只小鼠死亡。在预实验基础上,药物正式试验剂量设置如表3:
[0159]
表3组别剂量设置
[0160][0161][0162]
试验室环境:室温24
±
2℃,相对湿度60~70%。观察指标:药物(实施例1所制得的化合物i
‑
1)按上述剂量腹腔注射给药2次,上午下午各一次,记录各组小鼠中毒症状及死亡情况,死亡动物进行尸检;观察期为14天。结果表明:化合物i
‑
1对小鼠有一定毒性,较高剂量下给药后会引起小鼠死亡;腹腔注射给药的ld
50
值为186.5641(160.9764
‑
216.2190)mg/kg。死亡动物尸检均未发现脏器明显异常。
[0163]
体重变化如图3,与对照组相比,未见明显毒性反应。
[0164]
he染色结果如图4,实施例1所制得的化合物(i
‑
1)对心、肝、脾、肺、肾等主要器官未见明显毒性。
[0165]
(5)、化合物抗前列腺癌(du
‑
145)活性测定:
[0166]
药物为实施例1所制得的化合物(i
‑
1);细胞株为人前列腺癌du
‑
145细胞;受试动物为spf级balb/c裸小鼠;受试动物为spf级balb/c裸小鼠;雄性;模型组8只,实验组各12只,共32只;药物剂量设置如表4。
[0167]
表4药物剂量配置
[0168][0169]
实验方法:收集培养的人前列腺癌du
‑
145细胞悬液,浓度为1x107个/ml,以每只0.1ml接种于裸小鼠右侧腋窝皮下;裸鼠移植瘤用游标卡尺测量移植瘤直径,接种23天后,肿瘤生长至70
‑
100mm3时将动物随机分组;同时,各组裸鼠开始给药,给药方案见组别与给药方案,使用测量瘤径的方法,动态观察受试样品的抗肿瘤效应。实验结束后,随即处死裸鼠,手术剥取瘤块称重。
[0170]
实验结果见表5所示:受试药实施例1对肿瘤生长有明显抑制作用,且高剂量组(20mg/kg)的抑制作用优于低剂量组(10mg/kg),高低剂量组的抑瘤率分别为83.2%与
68.5%。加药组与对照组相比,对动物的体重无明显影响。具体肿瘤生长曲线见图5。
[0171]
因此,实施例1制得的受试药(i
‑
1)对人前列腺癌du
‑
145裸鼠异种移植瘤生长有明显的抑制作用,对动物的体重无明显影响,可作为adar1靶点相关疾病的候选治疗化合物。
[0172]
表5受试样品对人前列腺癌细胞du
‑
145裸鼠移植瘤肿瘤生长的影响
[0173][0174]
与模型对照组比较,
*
p<0.05,
**
p<0.01
[0175][0176]
与模型对照组比较,
*
p<0.05,
**
p<0.01
[0177]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。