1.本发明属于药物技术领域,涉及药物杂质检测分析领域,具体涉及一种替莫唑胺原料药中甲胺含量的测定方法。
背景技术:
2.替莫唑胺(temozolomide)为一种烷化剂型抗癌药物,由英国can research ventures公司首创,schering-plough公司开发,1999年8月经fda批准在美国获准上市,同时在欧洲多国上市。具有广谱抗肿瘤活性[l.h.tsang,et al.cancer chemother pharmacol.27(1991):342-346],尤其是对神经胶质瘤(脑癌)和黑色素瘤(皮肤癌)。替莫唑胺的胶囊剂已在欧美批准用于治疗恶性神经胶质瘤。
[0003]
药品的安全有效是当前人们普遍关注的重要问题,现有的替莫唑胺合成工艺以5-氨基-咪唑-4-甲酰胺或其盐酸盐为原料,经重氮化合成5-重氮基-咪唑-4-甲酰胺,然后在极性溶剂dmso存在下与异氰酸甲酯反应合成替莫唑胺。原料5-氨基-咪唑-4-甲酰胺的合成工艺如下:
[0004][0005]
在原料5-氨基-咪唑-4-甲酰胺的合成过程中使用了甲酰胺,甲酰胺的合成工艺如下:
[0006][0007]
因此,替莫唑胺原料药中有存在甲胺的可能,甲胺属有毒物质。按照ich m7指导原则规定,替莫唑胺属抗癌药,因此根据其服药周期1-10年,得出mic的每日最大摄入限量可按照10μg计算,替莫唑胺日最大服用剂量按照400mg计算,则限度相当于25ppm(0.0025%),因此,替莫唑胺中甲胺限度要求不超过25ppm(0.0025%)。然而,我国在替莫唑胺的法定标准中,尚未收载替莫唑胺中有甲胺的检查方法,标准缺失,国内外均无相关文献报道,无法评价药品质量,存在安全隐患。
技术实现要素:
[0008]
针对现有技术的不足,本发明的目的在于提供一种检测替莫唑胺原料药中甲胺的方法,该方法具有过程简单、使用成本低廉、精密度好、准确度高等特点,能快速地测定替莫
唑胺原料药中甲胺的含量。
[0009]
本发明是通过以下技术方案实现的:
[0010]
替莫唑胺原料药中甲胺含量的测定方法,包括以下步骤:
[0011]
配制对照品溶液:精密称定甲胺盐酸盐,加乙腈、衍生化试剂、硼酸盐缓冲溶液稀释制备对照品溶液;
[0012]
配制供试品溶液:精密称定替莫唑胺原料药,加乙腈、衍生化试剂、硼酸盐缓冲溶液稀释制备供试品溶液;
[0013]
配制空白溶液:精密量取乙腈、衍生化试剂、硼酸缓冲溶液、氨水1ml,用乙腈稀释配制空白溶液;
[0014]
采用高效液相色谱法分析,精密量取空白溶液、供试品溶液与对照品溶液,分别注入液相色谱仪,记录色谱图;对比空白溶液色谱图和对照品溶液色谱图,确定甲胺衍生物峰,供试品溶液的色谱图中如有与对照品溶液色谱图中甲胺衍生物峰保留时间一致的峰,按外标法以峰面积计算供试品中甲胺盐酸盐的含量,所述甲胺盐酸盐的含量乘以换算系数0.46,即得替莫唑胺原料药中甲胺的含量。
[0015]
本发明的进一步改进方案为:
[0016]
所述对照品溶液溶液的配制过程为:精密称定甲胺盐酸盐约20mg~25mg,置100ml量瓶中,加水1ml使溶解,加乙腈稀释至刻度,摇匀;精密量取1ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀;精密量取1ml,置20ml量瓶中,加乙腈10ml,加衍生化试剂1ml、硼酸盐缓冲溶液1ml,振摇5分钟,加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为对照品溶液。
[0017]
进一步的,所述供试品溶液的配制过程为:精密称定替莫唑胺原料药9mg~11mg,置20ml量瓶中,加乙腈10ml,超声使溶解,加衍生化试剂1ml、硼酸缓冲溶液1ml,振摇5分钟,加氨水1ml,摇匀,用乙腈稀释至刻度,摇匀,作为供试品溶液。
[0018]
进一步的,所述空白溶液的配制过程为:精密量取乙腈10ml,置20ml量瓶中,加衍生化试剂1ml、硼酸缓冲溶液1ml,振摇5分钟,加氨水1ml,摇匀,用乙腈稀释至刻度,摇匀,作为空白溶液。
[0019]
进一步的,所述衍生化试剂的配制过程为:精密称取氯甲酸-9-芴甲酯45mg~55mg,置50ml量瓶中,加乙腈溶解并稀释至刻度,摇匀,作为衍生化试剂.
[0020]
进一步的,所述硼酸缓冲溶液的配制过程为:称取硼砂适量,加水溶解,配制成ph为9.18的溶液,作为硼酸缓冲溶液。
[0021]
进一步的,所述高效液相色谱法的色谱条件为:用十八烷基硅烷键合硅胶为填充剂(welch ultimate xb-c18,4.6mm
×
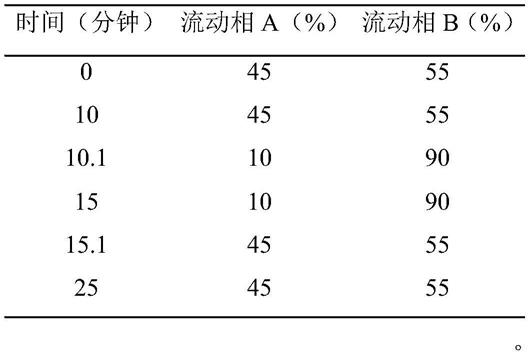
150mm,5μm);以0.1%甲酸溶液为流动相a,乙腈为流动相b,梯度洗脱;柱温为30℃;流速为每分钟1.0ml;检测波长为265nm进样量为10μl,所述梯度洗脱的过程为:
[0022][0023]
本发明的有益效果为:
[0024]
本发明所提供的方法,采用液相色谱进行分离与检测,设备普及率高,使用成本低廉,从而可以有效地扩大本方法的适用场合及使用范围。
[0025]
本发明所提供的检测方法中,所用试剂均无污染,对照品和供试品的处理过程简单,且对照品和供试品的使用量小,可以方便、快速地配制成对照品溶液、供试品溶液以及自身对照溶液。
[0026]
在本发明所提供的色谱条件下,杂质与主成分之间分离度良好,系统稳定性、重复性良好,准确度高,出峰时间快,分析时间短,节约实验成本。
[0027]
本发明所提供的方法,可快速、有效、简便对替莫唑胺中杂质量进行测定,既可以用于对药品成分进行评价或评估,避免存在安全隐患,又可以为该原料药的质量安全提供可靠的数据监测。
[0028]
利用试验对本发明所提供的检测方法进行了验证,结果表明,本方法的专属性强、精密度好,准确度高,能快速地测定替莫唑胺原料药中甲胺的量。
附图说明
[0029]
图1为实施例1所得的谱图;
[0030]
图2为实施例2所得的谱图;
[0031]
图3为实施例3所得的谱图;
[0032]
图4为甲胺的标准曲线;
[0033]
由于篇幅有限,方法验证部分的谱图没有在说明书中记载。
具体实施方式
[0034]
实验用仪器及色谱柱:
[0035]
仪器:
[0036]
仪器1:shimadzu lc-20a编号:jq1320;
[0037]
仪器2:shimadzu lc-20a编号:jq1321;
[0038]
色谱柱:
[0039]
色谱柱1:welch ultimate xb-c184.6mm
×
150mm,5μm),sn:60180700849,ln:2101.105;
[0040]
色谱柱1:welch ultimate xb-c184.6mm
×
150mm,5μm),sn:60180501645,ln:2101.105;
[0041]
电子天平:sartorius ms125p,编号:jb3114;
[0042]
实验用试剂:
[0043][0044]
实施例1~实施例3
[0045]
色谱条件:
[0046]
用十八烷基硅烷键合硅胶为填充剂(welch ultimate xb-c18,4.6mm
×
150mm,5μm);以0.1%甲酸溶液为流动相a,乙腈为流动相b,梯度洗脱;柱温为30℃;流速为每分钟1.0ml;检测波长为265nm进样量为10μl,所述梯度洗脱的过程为:
[0047][0048]
衍生化试剂的配制:精密称取氯甲酸-9-芴甲酯45mg~55mg,置50ml量瓶中,加乙腈溶解并稀释至刻度,摇匀,作为衍生化试剂.
[0049]
所述硼酸缓冲溶液的配制:称取硼砂适量,加水溶解,配制成ph为9.18的溶液,作为硼酸缓冲溶液。
[0050]
对照品溶液溶液的配制:精密称定甲胺盐酸盐约20mg~25mg,置100ml量瓶中,加水1ml使溶解,加乙腈稀释至刻度,摇匀;精密量取1ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀;精密量取1ml,置20ml量瓶中,加乙腈10ml,加衍生化试剂1ml、硼酸盐缓冲溶液1ml,振摇5分钟,加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为对照品溶液。
[0051]
供试品溶液的配制:精密称定替莫唑胺原料药9mg~11mg,置20ml量瓶中,加乙腈10ml,超声使溶解,加衍生化试剂1ml、硼酸缓冲溶液1ml,振摇5分钟,加氨水1ml,摇匀,用乙腈稀释至刻度,摇匀,作为供试品溶液。
[0052]
空白溶液的配制:精密量取乙腈10ml,置20ml量瓶中,加衍生化试剂1ml、硼酸缓冲溶液1ml,振摇5分钟,加氨水1ml,摇匀,用乙腈稀释至刻度,摇匀,作为空白溶液。
[0053]
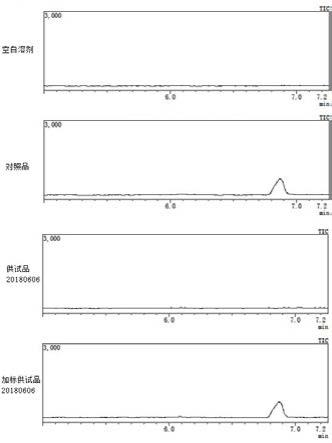
精密量取空白溶液、供试品溶液与对照品溶液各10μl,分别注入液相色谱仪,记录色谱图。对比空白溶液色谱图和对照品溶液色谱图,确定甲胺衍生物峰。供试品溶液的色谱图中如有与对照品溶液色谱图中甲胺衍生物峰保留时间一致的峰,按外标法以峰面积计算供试品中甲胺盐酸盐的含量,所述甲胺盐酸盐的含量乘以换算系数0.46,即得替莫唑胺原料药中甲胺的含量。
[0054]
取不同批次供试品溶液进样分析,所得谱图见图1至图3,计算甲胺的含量,结果见下表。
[0055]
表1样品中甲胺的测定结果
[0056]
序号批号甲胺(≤0.1%)实施例120191201未检出实施例220191202未检出实施例320191203未检出
[0057]
实验结果表明,三个批次样品中均未检出甲胺。
[0058]
分析方法验证
[0059]
1、专属性
[0060]
(1)空白溶液:精密量取乙腈10ml,置20ml量瓶中,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为空白溶液。
[0061]
(2)对照品贮备溶液:取甲胺盐酸盐约22mg(甲胺与甲胺盐酸盐的换算系数为0.46),精密称定,置100ml量瓶中,加水1ml使溶解,加乙腈稀释至刻度,摇匀;精密量取1ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀,作为对照品贮备溶液;
[0062]
(3)对照品溶液:精密量取对照品贮备溶液1ml,置20ml量瓶中,加乙腈10ml,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为对照品溶液。
[0063]
(4)供试品溶液:精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为供试品溶液。
[0064]
(5)供试品加标溶液:精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入对照品贮备液、衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为供试品加标溶液。
[0065]
精密量取上述溶液各10μl,分别注入液相色谱仪,记录色谱图,考察甲胺衍生物峰系统适用性,结果见表2
[0066]
表2,甲胺衍生物峰系统适用性考察结果
[0067]
溶液保留时间/min与相邻峰的分离度理论塔板数拖尾因子对照品溶液5.9846.9121721.04供试品溶液5.9706.9121881.06供试品加标溶液5.9626.8121381.04
[0068]
结果表明:对比空白溶液色谱图和对照品溶液色谱图,确定甲胺衍生物峰;空白溶
液、供试品溶液对甲胺衍生物峰测定无干扰。甲胺衍生物峰的拖尾因子、理论板数均符合规定;甲胺衍生物峰与相邻杂质峰分离度良好;替莫唑胺原料药中甲胺含量测定方法专属性良好。
[0069]
2、检测限及定量限
[0070]
(1)定量限
[0071]
用信噪比法确定定量限,以信噪比为10:1时相应浓度确定定量限。精密量取线性项下100%线性溶液1ml,置20ml量瓶,用乙腈溶解并稀释至刻度,摇匀;精密量取上述溶液1ml,置20ml量瓶,用乙腈溶解并稀释至刻度,摇匀,作为甲胺定量限溶液,结果浓度为0.0013μg/ml,为限度的0.26%。
[0072]
结果表明,甲胺定量限溶液精密度良好。
[0073]
(2)检测限
[0074]
用信噪比法确定检测限,以信噪比为3:1时相应浓度确定检测限。精密量取甲胺定量限溶液3ml,置10ml量瓶,用乙腈稀释至刻度,摇匀,作为甲胺检测限溶液;结果浓度为0.00039μg/ml,为限度的0.078%。
[0075]
3、线性及范围
[0076]
取甲胺盐酸盐约22mg(甲胺与甲胺盐酸盐的换算系数为0.46),精密称定,置100ml量瓶中,加水1ml使溶解,加乙腈稀释至刻度,摇匀;精密量取1ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀,作为对照品贮备溶液;
[0077]
精密量取对照品贮备液0.2ml、0.5ml、0.8ml、1ml、1.2ml、2ml,分别置20ml量瓶中,加乙腈10ml,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,分别作为20%、50%、80%、100%、120%、200%线性溶液。
[0078]
分别精密量取上述线性溶液及定量限溶液各10μl,注入液相色谱仪,结果表3,甲胺的标准曲线见图4。
[0079]
表3,甲胺线性测定结果
[0080]
/浓度(μg/ml)峰面积loq0.001350320%0.10453505550%0.26138794480%0.4181143109100%0.5226177110120%0.6271215921200%1.0451361532
[0081]
结果表明:甲胺在0.0013μg/ml~1.0451μg/ml的浓度范围内,线性关系良好。回归方程:y=346169x-1485.6,相关系数:r2=0.9999。
[0082]
4、精密度
[0083]
(1)仪器精密度
[0084]
精密量取对照品溶液10μl,重复进样6次,计算甲胺衍生物峰面积及保留时间的rsd(%),结果表4、表5:
[0085]
表4,仪器精密度测定结果(jq1320)
[0086][0087][0088]
表5,仪器精密度测定结果(jq1321)
[0089]
精密度峰面积保留时间/min11782246.10121780466.10931778346.10941776266.11351772286.11161765326.113平均值1775826.109sd6200.004rsd%0.350.07
[0090]
结果表明:本方法的仪器精密度良好。
[0091]
(2)重复性
[0092]
取甲胺盐酸盐约22mg(甲胺与甲胺盐酸盐的换算系数为0.46),精密称定,置100ml量瓶中,加水1ml使溶解,加乙腈稀释至刻度,摇匀;精密量取1ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀,作为对照品贮备溶液;
[0093]
精密量取对照品贮备溶液1ml,置20ml量瓶中,加乙腈10ml,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为对照品溶液。
[0094]
精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为供试品溶液。
[0095]
精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入对照品贮备液、衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为供试品加标溶液(平行配制6份)。
[0096]
精密量取对照品溶液、供试品溶液及供试品加标溶液各10μl,计算甲胺回收率及
含量rsd(%),结果见下表:
[0097]
表6,重复性测定结果
[0098][0099][0100]
结果表明:本方法的重复性良好。
[0101]
(3)中间精密度
[0102]
分别配制对照品溶液、供试品溶液及供试品加标溶液,配制方法同重复性项下。
[0103]
考察不同仪器,不同人员、不同日期的供试品加标溶液中甲胺回收率及含量的rsd(%),结果见下表:
[0104]
表7,中间精密度测定结果
[0105][0106]
结果表明:本方法的中间精密度良好。
[0107]
4、准确度
[0108]
取甲胺盐酸盐约22mg(甲胺与甲胺盐酸盐的换算系数为0.46),精密称定,置100ml量瓶中,加水1ml使溶解,加乙腈稀释至刻度,摇匀;精密量取1ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀,作为对照品贮备溶液;
[0109]
精密量取对照品贮备溶液1ml,置20ml量瓶中,加乙腈10ml,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为对照品溶液。
[0110]
精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为供试品溶液。
[0111]
采用标准加入法,精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入对照品贮备液0.2ml、衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为20%回收率溶液(平行配制3份);
[0112]
采用标准加入法,精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入对照品贮备液0.8ml、衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为80%回收率溶液(平行配制3份);
[0113]
采用标准加入法,精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入对照品贮备液1ml、衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为100%回收率溶液(平行配制3份);
[0114]
采用标准加入法,精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入对照品贮备液1.2ml、衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为120%回收率溶液(平行配制3份)。
[0115]
精密量取对照品溶液、供试品溶液、20%回收率溶液、80%回收率溶液、100%回收率溶液及120%回收率溶液各10μl,分别注入液相色谱仪,记录色谱图,计算甲胺回收率rsd(%)。
[0116]
结果见下表:
[0117]
表8,准确度测定结果
[0118]
[0119][0120]
结果表明:本方法准确度良好
[0121]
5、耐用性
[0122]
取甲胺盐酸盐约22mg(甲胺与甲胺盐酸盐的换算系数为0.46),精密称定,置100ml量瓶中,加水1ml使溶解,加乙腈稀释至刻度,摇匀;精密量取1ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀,作为对照品贮备溶液;
[0123]
精密量取对照品贮备溶液1ml,置20ml量瓶中,加乙腈10ml,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为对照品溶液。
[0124]
精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为供试品溶液。
[0125]
精密称取替莫唑胺(批号:20191201)约10mg,置20ml量瓶中,加乙腈10ml,超声使溶解,精密加入对照品贮备液、衍生化试剂及硼酸盐缓冲液各1ml,振摇5分钟,再加氨水1ml,摇匀,加乙腈稀释至刻度,摇匀,作为供试品加标溶液。
[0126]
通过改变流速(1.0
±
0.1ml/min)、柱温(30
±
2℃)、检测波长(265
±
2℃)和柱温(初温40
±
2℃,终温150
±
2℃),分别测定供试品加标溶液中甲胺回收率及含量rsd(%),结果见下表:
[0127]
表9,耐用性测定结果
[0128][0129][0130]
结果表明:本方法的耐用性良好。
[0131]
6、溶液稳定性
[0132]
分别配制对照品溶液及供试品加标溶液各一份,配制方法同专属性项下。
[0133]
精密量取对照品溶液及供试品加标溶液各10μl,连续12小时内,间隔1h分别注入液相色谱仪,记录色谱图,计算甲胺衍生物峰面积的rsd(%)。结果表10、表11:
[0134]
表10,溶液稳定性测定结果(对照品溶液)
[0135][0136]
表11,溶液稳定性测定结果(供试品加标溶液)
[0137]
[0138][0139]
结果表明:对照品溶液、供试品加标溶液在12小时内稳定性良好。
[0140]
甲胺含量测定方法学验证总结
[0141]
表12,甲胺含量测定方法学验证总结
[0142]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。