一种山羊亲子鉴定snp分子标记及其应用
技术领域
1.本发明涉及动物遗传育种领域,特别涉及一种山羊亲子鉴定snp分子标记及其应用。
背景技术:
2.内蒙古绒山羊是经长期自然选择和人工选育而成的特色优良畜种,因产绒量高、绒毛品质好、遗传性能稳定而闻名于世,是我国畜牧业中特色畜种之一。然而在养殖过程中,存在系谱记录错误的情况,会直接影响育种值估计和个体选择的准确性,极大地限制了我国绒山羊的遗传改良进程。
3.亲子鉴定是涉及医学、遗传学等相关学科的一门技术,通过分析被测个体与疑似亲本间的相似程度,来判断两者是否存在亲子关系。依据孟德尔遗传定律,在配子细胞形成前染色体成对分布在体细胞内,体细胞进行减数分裂,使染色体分离进入不同的配子中——每个配子只获得亲本(父或母)的一半遗传因子,精卵细胞结合形成合子,最终发育形成子代。因此,父母代都将一半的遗传因子传递给后代,定会遵循孟德尔遗传定律,倘若疑似亲本与子代有相同的遗传因子,则不排除此个体是子代亲本的可能性,若疑似亲本不包含子代的遗传因子,则此个体不可能是子代的亲本。
4.亲子关系分析旨在使用等显性分子标记识别个体间关系。被广泛应用于动物学、生态学和农业研究等领域。亲子关系分析的常见做法是通过逐个排除非亲缘个体或基于似然概率匹配最似亲缘个体来实现。尽管在过去的三十年中,使用的分子标记已从等位酶、微卫星(str)逐步更新为snp,但亲子分析的理论基础并未偏离遵循孟德尔定律的要求。
5.snp是指在基因组水平由于单个碱基的颠换(嘧啶和嘌呤之间的颠换)、转换(嘌呤间转换或嘧啶间转换)、缺失或插入等现象造成了dna序列多态性。颠换与转换是snp变异的常见形式,且发生概率后者是前者的2倍。由于胞嘧啶常因甲基化而脱去氨基发生转换,故snp多发生为c转换为t。通常来讲,snp标记的最小等位基因频率在群体中不小于1%,但不排除有小于1%的情况存在(如cdna)。
6.snp标记具有以下特点:(a)遗传稳定性高(突变率低):snp是基于单个碱基的突变,在不同生物中,突变频率约在10
‑9至5
×
10
‑9之间,而str(是由1
‑
6碱基为重复单位的短串联重复序列)的突变频率约在10
‑6至10
‑2之间,故snp遗传稳定性相对较高。(b)位点丰富、覆盖密度大:据研究表明,在哺乳动物中每500
‑
1,000bp就会出现一个snp,在人的基因组中平均每1,000bp有一个snp。但基因组中平均每15,000bp才有一个str。(c)分型准确且可自动化:目前为止,str分型是通过page或毛细管电泳两种方法,步骤繁琐且由于影子带和杂峰的存在,还需人工矫正易出现分型错误。而snp可通过pcr、杂交荧光检测、光谱或电子信号以及高通量测序等方法进行检测,不仅方法多样且相对准确易实现自动化。(d)具有代表性:snp大部分位于非编码区中,少部分在基因组编码区的snp有导致蛋白质功能改变的可能,这可能是直接造成生物体发生变异或病变的原因,因此可为遗传机理研究提供参考。随着对snp标记研究的不断加深,snp有望成为最常用的分子标记。
技术实现要素:
7.本发明的目的是提供一种山羊亲子鉴定snps分子标记,以及该snps分子标记在山羊亲子鉴定、育种领域、系谱制定领域、系谱校验领域、个体间遗传关系领域的应用。
8.本发明的一个方面,提供了一种山羊亲子鉴定snp分子标记,该snp分子标记包括:位于山羊三代基因组ars1版本1号染色体上第35315249bp处的g/c突变,其核苷酸序列如seq id no:1所示,其中序列中的m显示碱基突变位置,其中m表示g或c;位于山羊三代基因组ars1版本1号染色体上第115934804bp处的g/a突变,其核苷酸序列如seq id no:2所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本2号染色体上第1214172bp处的a/g突变,其核苷酸序列如seq id no:3所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本2号染色体上第47541960bp处的g/a突变,其核苷酸序列如seq id no:4所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本2号染色体上第57915126bp处的a/g突变,其核苷酸序列如seq id no:5所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本2号染色体上第85640919bp处的a/g突变,其核苷酸序列如seq id no:6所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本3号染色体上第12238669bp处的g/a突变,其核苷酸序列如seq id no:7所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本3号染色体上第23100139bp处的a/g突变,其核苷酸序列如seq id no:8所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本3号染色体上第36346016bp处的g/a突变,其核苷酸序列如seq id no:9所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本3号染色体上第66946532bp处的a/g突变,其核苷酸序列如seq id no:10所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本4号染色体上第3721258bp处的a/g突变,其核苷酸序列如seq id no:11所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本4号染色体上第46769604bp处的a/c突变,其核苷酸序列如seq id no:12所示,其中序列中的m显示碱基突变位置,其中m表示a或c;位于山羊三代基因组ars1版本4号染色体上第115273960bp处的g/a突变,其核苷酸序列如seq id no:13所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本5号染色体上第106137302bp处的g/a突变,其核苷酸序列如seq id no:14所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本5号染色体上第117814863bp处的a/g突变,其核苷酸序列如seq id no:15所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本6号染色体上第8836677bp处的a/g突变,其核苷酸序列如seq id no:16所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本6号染色体上第22425512bp处的c/g突变,其核苷酸序列如seq id no:17所示,其中序列中的m显示碱基突变位置,其中m表示c或g;位于山羊三代基因组ars1版本6号染色体上第89326703bp处的a/g突变,其核苷酸序列如seq id no:18所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本8号染色体上第42979080bp处的a/g突变,其核苷酸序列如seq id no:19所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本8号染色体上第68203190bp处的a/g突变,其核苷酸序列如seq id no:20所
示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本9号染色体上第42704426bp处的g/a突变,其核苷酸序列如seq id no:21所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本9号染色体上第83736477bp处的c/g突变,其核苷酸序列如seq id no:22所示,其中序列中的m显示碱基突变位置,其中m表示c或g;位于山羊三代基因组ars1版本10号染色体上第23837730bp处的a/g突变,其核苷酸序列如seq id no:23所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本10号染色体上第90325228bp处的g/a突变,其核苷酸序列如seq id no:24所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本11号染色体上第21931217bp处的a/g突变,其核苷酸序列如seq id no:25所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本11号染色体上第85327779bp处的g/a突变,其核苷酸序列如seq id no:26所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本12号染色体上第32658191bp处的g/a突变,其核苷酸序列如seq id no:27所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本12号染色体上第60926299bp处的a/g突变,其核苷酸序列如seq id no:28所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本12号染色体上第75210084bp处的a/c突变,其核苷酸序列如seq id no:29所示,其中序列中的m显示碱基突变位置,其中m表示a或c;位于山羊三代基因组ars1版本13号染色体上第23113494bp处的a/g突变,其核苷酸序列如seq id no:30所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本13号染色体上第58411086bp处的a/g突变,其核苷酸序列如seq id no:31所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本14号染色体上第1889687bp处的a/g突变,其核苷酸序列如seq id no:32所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本14号染色体上第17549473bp处的g/a突变,其核苷酸序列如seq id no:33所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本14号染色体上第52284360bp处的g/a突变,其核苷酸序列如seq id no:34所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本15号染色体上第67482760bp处的g/a突变,其核苷酸序列如seq id no:35所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本16号染色体上第29855092bp处的g/a突变,其核苷酸序列如seq id no:36所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本17号染色体上第1530981bp处的g/a突变,其核苷酸序列如seq id no:37所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本17号染色体上第40790363bp处的g/a突变,其核苷酸序列如seq id no:38所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本18号染色体上第64587854bp处的g/a突变,其核苷酸序列如seq id no:39所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本20号染色体上第2228968bp处的a/g突变,其核苷酸序列如seq id no:40所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本20号染色体上第39618012bp处的a/g突变,其核苷酸序列如seq id no:41所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本23号染色体上第
13434370bp处的a/g突变,其核苷酸序列如seq id no:42所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本24号染色体上第43142765bp处的a/g突变,其核苷酸序列如seq id no:43所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本25号染色体上第1400734bp处的g/a突变,其核苷酸序列如seq id no:44所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本26号染色体上第27643092bp处的a/g突变,其核苷酸序列如seq id no:45所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本26号染色体上第42218759bp处的g/a突变,其核苷酸序列如seq id no:46所示,其中序列中的m显示碱基突变位置,其中m表示g或a;位于山羊三代基因组ars1版本27号染色体上第22005018bp处的a/g突变,其核苷酸序列如seq id no:47所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本27号染色体上第42367823bp处的a/g突变,其核苷酸序列如seq id no:48所示,其中序列中的m显示碱基突变位置,其中m表示a或g;位于山羊三代基因组ars1版本28号染色体上第507372bp处的c/a突变,其核苷酸序列如seq id no:49所示,其中序列中的m显示碱基突变位置,其中m表示c或a;位于山羊三代基因组ars1版本29号染色体上第31461515bp处的a/g突变,其核苷酸序列如seq id no:50所示,其中序列中的m显示碱基突变位置,其中m表示a或g;选择其中1个或者多个snp分子标记的组合来用于山羊亲子鉴定。
9.在某实施方式中,该山羊品种为内蒙古绒山羊。
10.本发明的另一个方面,提供了一种上述snp分子标记在山羊亲子鉴定上的应用。
11.在某实施方式中,上述snp分子标记应用在内蒙古绒山羊的亲子鉴定上。
12.本发明的第三个方面,提供了一种snp分子标记应用于山羊亲子鉴定的方法包括:
13.对待鉴定对象进行snp基因型分型,所述的snp分子标记如权利要求1所示;
14.对待测个体基因型分型数据进行生物信息学分析,包括质量控制、pca分析以及贪婪算法,筛选适用于山羊亲子鉴定的snps组合;
15.根据待测个体在最终筛选的snps分子标记的基因型数据,使用cervus3.0.7软件基于似然法计算出lod和delta值,根据lod和delta值进行亲子关系推断。
16.在某实施方式中,该亲子鉴定方法中当lod值大于0时,候选亲本有可能是真实亲代,lod值最高的个体是最似亲本;当lod值小于0时,候选亲本不可能是真实亲本。当delta大于delta临界值时,候选亲本可能为真实亲本;当delta值等于0时,无法推断候选亲本的真实性。本发明的第四个方面,提供了一种snp分子标记在山羊育种领域的应用。
17.本发明的第五个方面,提供了一种snp分子标记在制定山羊系谱领域的应用。
18.本发明的第六个方面,提供了一种snp分子标记在校验山羊纸质系谱领域的应用。
19.本发明的第七个方面,提供了一种snp分子标记在确定个体间遗传关系领域的应用。
20.本发明的有益效果:
21.(1)对芯片测序数据进行处理后,初步获得124个snps分子标记,通过多态性分析发现内蒙古绒山羊群体存在丰富的遗传多态性,具有很大的育种价值和性状改良潜力。snp位点的pic介于0.25
‑
0.5之间,属于中度多态,且全部位点不偏离哈代
‑
温伯格平衡,说明位点无分型问题,可以用于亲子关系分析。
22.(2)通过对不同梯度组合进行模拟分析发现:累积排除率与标记数目成正比,当位点增大到一定数量,其值将保持不变,最高为1,说明snp位点过多可能会造成资金的浪费,反而对鉴定无利。当snp位点数量为50时,能达到较高的累积排除率(cpe1、cpe2、cpe3分别超过了99.87%、99.99%、99.99%),并且能够与初步获得的124个snps分子标记达到同等的推断率(100%),说明这50个snps分子标记可替代124个分子标记进行后续亲子鉴定研究。
23.(3)使用50个snps对有明确父子、母子关系的个体进行鉴定,其结果与系谱记录全部一致,表明筛选的这50个snps分子标记具有亲子鉴定效力。并对有纸质系谱记载的182对父子及163对母子进行鉴定,其结果与系谱记录的一致率为80.78%和80.98%,说明系谱完整性有待进一步提高,通过采用这些snps分子标记进行亲子鉴定可以校验纸质系谱的不足,为制定正确的系谱提供科学依据,进一步对于该物种的育种值计算、遗传育种工作提供保障。该snps分子标记也可以应用于个体间遗传关系确定、大规模种群的亲子关系确定、家系系谱创建等领域。
附图说明
24.图1选用的内蒙古绒山羊出生年份分布图;
25.图2为初步质控后各染色体上snps分布图。
具体实施方式
26.下面结合附图及实施例对发明作进一步详细的说明。
27.实施例一亲子鉴定snp分子标记的筛选
28.1.1材料与方法
29.1.1.1数据来源
30.基于课题组前期内蒙古绒山羊ggp_goat_70k芯片(后文均用70k芯片表示)(详见内蒙古农业大学博士学位论文“山羊snp芯片设计与内蒙古绒山羊重要经济性状的全基因组关联分析和基因组选择研究”)测序数据,数据共包含1,880个体(全部为内蒙古亿维白绒山羊有限责任公司的阿尔巴斯型内蒙古绒山羊),其中有2010年出生的15只,2011年出生的152只,2012年出生的184只,2013年出生的有258只,2014年出生的226只,2015年出生的235只,2016年出生的254只,2017年出生的460只,2018年出生的96只(如图1所示)。
31.1.1.2数据处理
32.(1)原始数据初步质控
33.该70k芯片共包含67,088个snps分子标记,首先利用plink软件对数据进行初步质控,质控条件如下:
34.①
snp分子标记位点位于常染色体上。
35.②
个体检出率大于90%。
36.③
每个标记的检出率大于90%。
37.④
次要等位基因频率(maf)大于0.01。
38.⑤
哈代——温伯格平衡检验(hw)p>1
×
10
‑6。
39.运行命令:plink
‑‑
file snp
‑‑
allow
‑
no
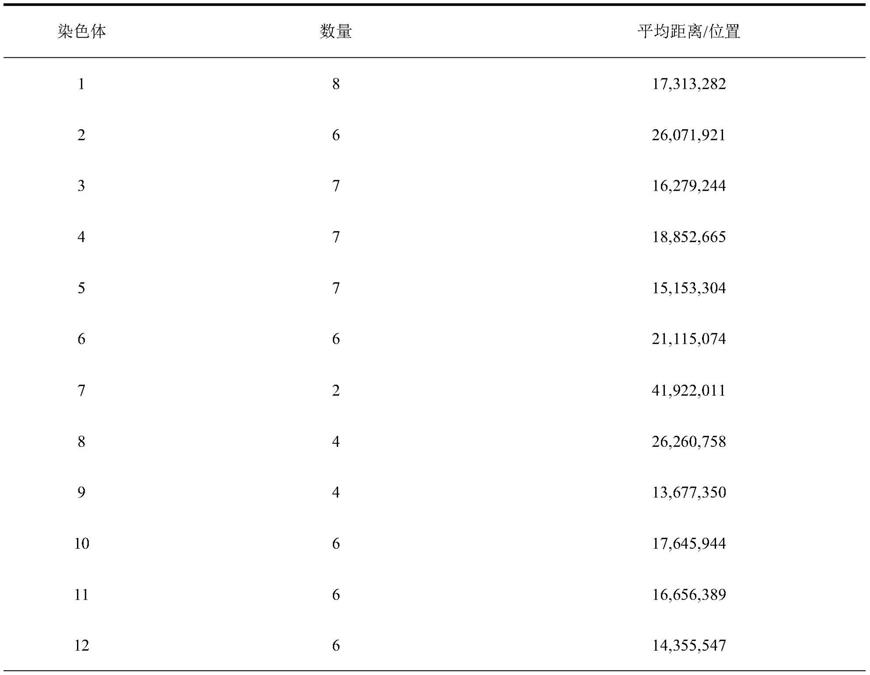
‑
sex
‑‑
mind 0.1
‑‑
geno 0.1
‑‑
maf0.01
‑‑
hwe 1e
‑6‑‑
recode 12
‑‑
allow
‑
extra
‑
chr
‑‑
chr
‑
set 29
‑‑
out file。
40.(2)数据填充
41.对质控后的数据进行格式转换,转换成由0,1,2组成的1,880行、53,066列的数据集(如:0表示基因型aa,1表示基因型aa,2表示基因型aa)。基于蒙特卡罗马尔科夫链(mcmc,markov chain monte carlo)算法,使用beagle软件对尚未分型成功的snp位点进行填充,用于后续分析。
42.(3)主成分分析(pca)
43.pca(principal componentsanalysis)是能够将较大变量集缩减为较小变量集,且信息损失较少的一种数据降维技术。使用rx644.0.2软件进行pca分析,对snp数据集进行降维,并保证snps之间能够相互独立。保留了累计贡献率达80%的主成分。
44.(4)进一步质控
45.首先,使用plink软件对每个主成分进一步严格质控,根据连锁不平衡分析去除强连锁位点,并去除不符合哈代
‑
温伯格平衡检验的位点(p<0.05),以及去除maf(minorallele frequency,次要等位基因频率)值小于0.3的snp位点。
46.(5)贪婪算法筛选snp
47.贪婪算法(greedy algorithm)是指在对问题进行求解时,能够在局部(不是对整体而言)做出最优选择的一种算法。通过cervus3.0.7软件对严格质控后的位点进行hexp(expected heterozygosity,期望杂合度)、pic(polymorphism information content,多态信息含量)、pe(power of exclusion,单个snp位点排除某疑似亲本的平均概率)等参数的计算。将每个位点的pe作为参考指标,采用贪婪算法对每个主成分的snp标记进行筛选,然后合并各个主成分所选snp标记,根据位点的pic高低并参考maf值进行位点剔除,并确保同一条染色体相邻snp之间的距离大于10mb,最后获得初步筛选的snp标记。其中:
48.①
等位基因频率(allele frequency)是指某一群体中某一特定等位基因在该基因座出现的次数占该基因组所有等位基因的比例。而maf是指群体中某一位点上不常见的等位基因发生的频率。等位基因频率可用于表示种群中基因的遗传多样性或者可以表示种群基因库的丰富度,计算公式如下:
49.p
i
=p 1/2∑h
i
ꢀꢀꢀꢀꢀꢀ
(1)
50.其中,pi为某一等位基因的频率;p为含某一等位基因的纯合子的频率;hi为含某一等位基因的杂合子的频率。
51.②
杂合度(heterozygosity)指的是一个特定的位点在一个群体中杂合子个体所占的比例,分为hobs(observed heterozygosity,观察杂合度)和hexp。hobs指所检测到杂合子个体占整个群体的比例,hexp指在某个或多个位点,一个个体为杂合子的预期概率,若hexp=0.5,则表示群体内的每个个体有50%的可能性成为杂合子,计算公式如下:
[0052][0053][0054]
其中pi为某一位点上第i个等位基因频率,n为此位点的等位基因数,ak为第k位点的杂合子个数,m为位点个数。
[0055]
③
pic(polymorphism information content)表示后代在其系谱结构中能提供的信息量,若pic=0.37,则说明37%的后代能为系谱结构的解释提供信息,计算公式如下:
[0056][0057]
其中pi和pj分别为群体中第i、j个等位基因频率,n为等位基因数。
[0058]
④
pe(probability ofexclusion)指排除某疑似亲本的平均概率。分为双亲未知(pe1)、单亲已知(pe2)和双亲已知(pe3)三种情况,计算公式如下:
[0059]
情况一:单亲未知时
[0060][0061]
情况二:单亲已知时
[0062][0063]
情况三:双亲已知时
[0064][0065]
其中,n为每个标记的等位基因个数,pi为第i个等位基因频率。
[0066]
⑤
cpe(cumulative probability ofexclusion,累积排除概率)为多个snp标记所累积的排除概率,计算公式如下:
[0067]
cpe=1
‑
(1
‑
p1)(1
‑
p2)(1
‑
p3)
…
(1
‑
p
l
)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(8)
[0068]
其中l为所选snp标记个数。
[0069]
1.1.3snp多态性研究及模拟分析
[0070]
对初步筛出的124个snps标记进行多态性分析,并按其pic高低划分9个snp梯度(各梯度snps标记相差10,均为124个snps中pic最高的),同时使用cervus3.0.7对9个不同梯度snp组合进行模拟分析,模拟参数为:模拟子代为10,000,候选亲本抽样率为1,位点分型率为1(依据等位基因频率结果),分型错误率设为0.01,95%为严格置信度,80%为宽松置信度。
[0071]
1.2结果
[0072]
1.2.1初步质控标记染色体分布
[0073]
对原始数据进行初步质控后,保留了53,066个snps标记,其在每条染色体上的分布见图2。
[0074]
1.2.2snp筛选结果
[0075]
经pca分析满足累计贡献率达80%的主成分共240个,经进一步质控、贪婪算法等筛选,初步获得124个snps位点满足条件。对124个snps位点进行统计发现每条染色体上有1
‑
8个不同数量的snps位点,且平均距离均大于12mb。21和25号染色体上仅存在一个位点,数量最少,1号染色体上存在8个位点,数量最多。29号染色体相邻位点间距最小,但也大于12mb。7号染色体相邻位点间距最大,超过了41mb,避免了位点间的连锁反应,有利于后续亲子鉴定的分析。(统计结果见表1)
[0076]
表1各染色体snp数目及snp平均间距
[0077]
[0078][0079]
1.2.3 124snps多态性研究结果
[0080]
对初步筛选的124个snps位点进行多态性分析,结果显示hobs在0.464
‑
0.534之间(0.5026
±
0.012),hexp在0.473
‑
0.500之间(0.4977
±
0.003),说明群体存在丰富的遗传多态性,具有很大的育种价值和性状改良潜力。pic在0.361
‑
0.375之间(0.3738
±
0.002)属中度多态。maf在0.488
‑
0.500之间(0.4965
±
0.003)。全部位点不偏离哈代
‑
温伯格平衡,表明无无效等位基因、分型偏差、位点连锁等问题,可以用于亲本分析,结果如表2所示。
[0081]
表2 124snps位点多态性信息
[0082]
[0083]
[0084]
[0085]
[0086]
[0087][0088]
注:locus为位点,hobs为观察杂合度,hexp为期望杂合度,pic为多态信息含量,maf为最小等位基因频率,hw为哈代
‑
温伯格平衡定律,ns表示不显著偏离
[0089]
另外,对124snps进行单个位点的排除概率(pe1、pe2、pe3)统计,并进行累积排除概率(cpe)的计算,发现三种鉴定类型都有很高的累计排除概率(均大于0.9999999),说明124个位点的亲子鉴定效果极好,结果如表3所示。
[0090]
表3 124snps单个位点排除率
[0091]
[0092]
[0093]
[0094]
[0095]
[0096][0097]
注:pe1为对一疑似亲本的平均排除概率,pe2为已知另一亲本基因型,对疑似亲本的平均排除概率,pe3为对疑似亲本对的平均排除概率
[0098]
1.2.4各梯度模拟结果
[0099]
对不同梯度snps组合进行模拟分析,20
‑
124snps模拟结果见表4
‑
表13,delta为子代两个第一和第二最似亲本的lod(likelihood ofodd,似然比的自然对数)差值,delta临界值越小(最小值为0)表明标记效果越好。由表4
‑
表12的delta临界值比较看出,临界值在95%的置信水平下由0.85下降到0,说明snps的鉴定效果越来越好。由表6可知,40个snps的鉴定效果良好,但是在分配率没有达到100%,可能会造成一定的误差,而50个snps后分配率均可以达到100%,因此将位点最终选为50个。
[0100]
表4 20snps位点模拟结果
[0101][0102]
表5 30snps位点模拟结果
[0103][0104]
表6 40snps位点模拟结果
[0105][0106]
表7 50snps位点模拟结果
[0107][0108]
表8 60snps位点模拟结果
[0109][0110][0111]
表9 70snps位点模拟结果
[0112]
[0113]
表10 80snps位点模拟结果
[0114][0115]
表11 90snps位点模拟结果
[0116][0117]
表12 100snps位点模拟结果
[0118][0119]
表13 124snps位点模拟结果
[0120][0121][0122]
对模拟结果进行统计分析(表14),发现cpe1由0.930791241(20snps)增大至0.999999924(124snps),cpe2由0.984084844(20snps)增大至1(124snps),cpe3由0.998636743(20snps)增大至1(70snps
‑
124 snps),且随位点增加排除率不变,说明cpe会随snp数目增加而增大,待位点增大到一定程度后,排除率保持不变最高为1。且随snps标记数目增加,50个以上snps标记的亲子分配率(80%、95%置信水平)都可达100%,且50个snps位点的cpe1达99.87%,cpe2和cpe3更是超过了99.99%,具有较好的排除效果,与124snps组合一样在两种置信水平下推断率能够达到100%。因此,最终选择这50个snps进行后续亲子鉴定研究。
[0123]
表14不同梯度组合排除概率及推断率
[0124][0125]
注:cpe1为对一疑似亲本的累积排除概率,cpe2为当另一亲本基因型已知时,对疑似亲本的累积排除概率,cpe3为对疑似亲本对的累积排除概率
[0126]
50个snps的详细信息见表15和表16。此组合的hobs的平均值为0.5056
±
0.0105,hexp的平均值为0.4998
±
0.0004,pic的平均值为0.375
±
0.0000,maf的平均值为0.4969
±
0.0027。
[0127]
表15 50个snps标记组合信息
[0128]
[0129]
[0130][0131]
表16 50个snps多态信息
[0132]
[0133]
[0134][0135]
注:locus为位点,hobs为观察杂合度,hexp为期望杂合度,pic为多态信息含量,maf为最小等位基因频率,hw为哈代
‑
温伯格平衡定律,ns表示不显著偏离
[0136]
选择亲子鉴定的taq snps时,首先要确保标记具有多态性,其次maf的值也是一个比较重要的参数,具有高maf的snp会在无关个体之间产生最高的相对纯合子频率,cpe随maf的增大而增大,且maf超过0.3时,cpe增加显著,其鉴定效果会更加准确。sanaranayp等在亲子鉴定研究中表明,snp标记的maf值对鉴定效果有很大的影响。van doormaal v等也指出maf值较小的snp亲子鉴定能力有限。王悦(2018)通过对影响亲子鉴定准确性因素的研究,确定了maf与snp数目是主要影响因素。周磊等指出一套最低maf>0.35的40个snps标记组合,可满足通常情况的亲子鉴定。fisher pj研究指出,平均maf达到0.35时,与14个str相比40个snps是更好的诊断工具。本研究最终筛选获得的50snps组合的maf范围在0.4883
‑
0.5之间,平均值约为0.4969
±
0.0027,符合满足maf>0.35的要求,因此,可用于常规亲子鉴定应用。
[0137]
在本研究中,50个snps标记的单一亲本鉴定累积排除概率大于99.87%,其他两种情况下的鉴定累计排除率则超过了99.99%。因此可以满足亲子鉴定需求,能够用于后续亲子鉴定的应用。
[0138]
实施例二snp标记用于内蒙古绒山羊亲子鉴定研究
[0139]
2.1材料与方法
[0140]
2.1.1试验数据
[0141]
(1)实施例一最终筛选的50个snps位点。
[0142]
(2)明确亲子关系的父子与母子各10对(共35个体)。
[0143]
(3)待测有系谱记录的163对母子(314个体)、182对父子(193个体)。
[0144]
2.1.2snp组合验证
[0145]
利用实施例一所筛选的50个snps位点,对具有明确亲子关系的10对父子和母子进行亲子鉴定,验证其snps组合的鉴定效力。
[0146]
2.1.3亲子对鉴定
[0147]
检测163对母子与182对父子是否存在亲子关系,与系谱记录比对,评估系谱记录的准确性。
[0148]
2.1.4cervus3.0.7软件分析
[0149]
(1)等位基因频率分析
[0150]
将个体基因型分型数据整理成cervus软件要求的逗号分隔格式(.csv),勾选标题行,依次填写对应的id栏、首个等位基因列和位点数。勾选哈代
‑
温伯格平衡测定框测定各位点是否符合哈代
‑
温伯格平衡其他选项默认。保存输出文件,设置好后点击ok。该分析能生成模拟亲本分析及亲本分析所需的等位基因频率,并计算各种汇总统计量,以评估位点用于亲本分析的适合性。
[0151]
(2)模拟亲本分析
[0152]
等位基因频率数据可选择等位基因频率输出的扩展名为.alf文件,勾选标题行,填写对应模拟参数(子代、候选亲本(对)、抽样比例、位点分型比例、分型错误率、最小分型位点),分型错误率为0.01。置信区间为80%和95%,最后保存输出文件点击ok。该分析用于估算位点的等位基因频率的解析能力,同时可以估计lod和delta临界值。
[0153]
(3)亲本分析
[0154]
创建子代及其已知亲本和疑似亲本的id文件(子代文件可包含亲本),选择适当亲本分析类型(母本、父本、亲本对性别已知或未知)。选择子代文件,勾选标题行选择子代id对应列,若子代文件包含亲本则需勾选对应选项并填写亲本id对应列,若不包含则点击next。候选亲本情况已经填写,继续next。选择对应个体基因型文件,勾选标题行填写对应id列及第一个等位基因列,选择扩展名为.alt文件,继续next。选择模拟分析结果输出的扩展名为.sim的文件,点击next。保存输出文件点击ok。该分析将为子代匹配最似的亲本。
[0155]
在cervus软件使用中,需要注意以下细节:
①
所用位点是否符合软件分析要求(位于常染色体、连锁平衡等)。
②
各输入文件(基因型文件、子代及亲本文件等)的信息是否一致、正确。
③
由于个体基因型缺失(基因型文件中没有该个体的基因型数据)和基因型被忽略(个体分型位点数小于模拟分析时设定的最小分型位点数)的原因,导致分型数据中无位点(一个或多个)的分型数据。
④
若以delta统计量进行亲本指派,可能会出现两个最似亲本的lod值相等,得出delta值为0。或者,以lod值作为统计量进行亲本指派,可能两个最似亲本lod值相等,最终无最可能匹配候选亲本。
⑤
对于另一亲本基因型已知,进行亲本(父本或母本)分析时,若已知亲本并非子代的真实亲本,则会误导候选亲本的指派。
⑥
若子代和亲本文件分为两个,在亲子分析步骤中,一定要注意文件是否有行标,子代和亲本所处列及亲本在文件中的出现方式(每代一行或所有子代一列)。
[0156]
2.1.5系谱比对
[0157]
将分析结果与系谱记录进行比对,系谱错误为两种情况:(1)cervus软件分析所得
lod值小于0或delta值小于临界值,说明该子代无匹配亲本即系谱记录错误。(2)子代经软件匹配亲本与系谱记录不一致,即说明系谱记录错误。
[0158]
2.2结果
[0159]
2.2.1snp组合的鉴定效力
[0160]
使用10对父子的鉴定结果对50个snps位点的鉴定效力进行验证(表17、表18)。由表17可知在严格与宽松置信水平下,delta临界值为0,鉴定效果极好,且被检测的10个子代全部获得最似父本,分配率达100%。表18显示所有子代与其候选亲本的错配位点都为0,且所有个体的lod值均大于0、delta值大于临界值,同时在95%的置信水平下均匹配了真实父本。
[0161]
表17 10对真实父子关系鉴定的参数
[0162][0163][0164]
表18 10对真实父子鉴定结果
[0165][0166]
注:配对置信度显示“*”为严格置信度(95%置信水平);“ ”为宽松置信度(80%置信水平);
“‑”
显示最可能候选亲本未被指派;如候选亲本不是最可能的,该列将为空白。
[0167]
由表19可知,同父子鉴定一样,被检测的10个子代全部获得最似母本,分配率达100%。表20显示除201701267号子代与其最似母本有1个错配位点外(在误差允许范围内),其他子代与最似母本的错配位点都为0,且所有个体的lod值均大于0、delta值大于临界值,同时在95%的置信水平下均匹配了真实母本。
[0168]
表19 10对真实母子关系鉴定的参数
[0169][0170]
表20 10对真实母子鉴定结果
[0171][0172]
注:;配对置信度显示“*”为严格置信度(95%置信水平);“ ”为宽松置信度(80%置信水平);
“‑”
显示最可能候选亲本未被指派;如候选亲本不是最可能的,该列将为空白。
[0173]
2.2.2利用snp检查系谱信息的准确性
[0174]
对芯片测序个体进行整理,共发现182对父子及163对母子存在系谱记录(不包含上述用于鉴定效力的10对父子和母子),因此通过这些个体来检查系谱记录的准确性。由于父
‑
子、母
‑
子鉴定个体过多,此处仅展示部分结果(结果如表21和22)。由父子鉴定结果可知,在严格和宽松置信度下delta临界值都为0,说明标记鉴定效力极佳,且都有162个子代匹配了最似父本,占总体的89%。其中有22对个体存在1
‑
6不等的错配位点,其中错配位点数大于1时,有201804144、201802181、201706298、201604150等20个子代的lod值小于0且delta值为0(不大于临界值),排除了最似亲本。除上述20个体外,其他子代均在95%的置信水平下匹配到最似父本。经系谱比对发现,另有201605005、201804262、201804163、201801166等15个个体的最似亲本与系谱记录亲本不一致。最终结果显示,在这182对父
‑
子中有147对与系谱一致,经计算一致率为80.77%。
[0175]
表21 182对父子鉴定分配参数
[0176][0177]
表22 182对父子鉴定结果
[0178]
[0179]
[0180]
[0181]
[0182]
[0183]
[0184]
[0185][0186]
注:配对置信度显示“*”为严格置信度(95%置信水平);“ ”为宽松置信度(80%置信水平);
“‑”
显示最可能候选亲本未被指派;如候选亲本不是最可能的,该列将为空白。
[0187]
由母子鉴定结果可知(如表23和24所示),在严格和宽松置信度下delta临界值分别为0.01和0,且都有158个子代获得母本指派,占总体的97%。由鉴定结果得知错配位点不高(为1
‑
3个),仅201702187、201606156、201703172、201805001、201605058这5个子代在严格置信水平下匹配的lod值小于0且delta值为0小于临界值0.01,排除了最似母本。除这5个子代外,其他个体均在95%置信水平下匹配到最似母本。经系谱比对发现,有26个子代的最似母本与系谱记录亲本不一致。最终结果3显示,在这163对母
‑
子中有132对与系谱一致,经计算一致率为80.98%。
[0188]
表23 163对母子鉴定分配参数
[0189][0190]
表24 163对母子鉴定结果
[0191]
[0192]
[0193]
[0194]
[0195]
[0196]
[0197][0198]
注:配对置信度显示“*”为严格置信度(95%置信水平);“ ”为宽松置信度(80%置信水平);
“‑”
显示最可能候选亲本未被指派;如候选亲本不是最可能的,该列将为空白。
[0199]
综上所述,系谱在遗传育种中至关重要,常规育种是建立在系谱和生产性能数据库基础上才能估计育种值,进而根据育种值的高低进行选种。目前较为流行的基因组选择虽然通过测定基因型可以得到亲缘关系矩阵,并进行育种值估计,但多数研究表明,使用最佳线性无偏预测法(gblup)估计的基因组育种值准确性要低于一步法(ssblup),主要是因为ssblup法可以有效的结合系谱数据,通过基于系谱的估算降低基因分型成本,减少育种值的基因组估计偏差,结合基因型和非基因型个体进行联合分析,增大测定群体数量,提高基因组育种值预测的准确性。当个体的父母没有被记录时,亲子分配算法可以使用遗传数据重建亲子关系。在过去的十年中,使用blup已在育种计划中实现重大遗传进展。而blup的关键在于使用加性遗传关系矩阵对育种值的估算,若系谱错误会将导致育种值估计准确性
降低。如此看来,在传统blup(ablup)中系谱相当于基石,没有系谱就无法完成遗传评估(遗传相关、育种值估计无法做到)。nwogwugwu cp通过比较基因组gblup和ssgblup对遗传评价的准确性,得出ssgblup的预测精度优于gblup方法。即说明使用ssblup在结合g阵与a阵后进行预测的准确性高于gblup,进一步说明系谱记录在育种实践中的重要性。
[0200]
在育种实践过程中,系谱错误能够对遗传改良、育种值估计等产生不利影响,进而降低对性状选择的准确性。有研究指出11%的父系错误率将导致群体遗传进展减少11%
‑
15%。亲子错误率达15%时,与无系谱错误相比,对于遗传力(h2)为0.2和0.5的性状,遗传进展分别下降16.9%和8.7%。经模拟发现对于h2为0.25的性状,10%的亲子关系错误率,将造成群体遗传进展降低4.3%,如果利用遗传标记对错误系谱进行纠正,可以带来很大的经济效应。系谱错误造成遗传进展下降的同时,还会对遗传相关、近交系数及公畜方差等的估计造成影响,而且对于基因组选择、qtl定位等利用系谱信息的相关研究也会造成不利影响。garc
í
a
‑
ruiz a等研究表明,基因分型和系谱恢复可能有助于提高种群的遗传改良效率。
[0201]
通过本研究结果,通过snp标记对内蒙古绒山羊的父子、母子鉴定,与系谱的一致率均在80%左右,说明在该群体中纸质系谱完整性有待进一步提高,后续拟对不一致的系谱信息进一步核对、加强系谱管理工作、避免系谱错误、保证其准确性,为相关育种工作提供可靠的数据保障,极致发挥系谱在育种实践中的作用。
[0202]
以上所述的仅是本发明的一些实施方式。对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。