1.本发明属于医药技术领域,具体涉及杂芳环类dna
‑
pk抑制剂化合物、其药学上可接受的盐或其异构体,含有所述化合物、其药学上可接受的盐或其异构体的药物组合物及制剂,制备所述化合物、其药学上可接受的盐或其异构体的方法,以及所述化合物、其药学上可接受的盐或其异构体的用途。
背景技术:
2.癌症是全世界难治疗的恶性疾病,治疗难度大,死亡率高,给患者和家庭带来沉重的负担,是影响我国居民健康的主要疾病。近年来,我国癌症发病率明显增加,其死亡率也呈逐渐上升趋势,癌症防治面临着严峻的形势。
3.目前,放疗以及化疗是除手术切除外治疗癌症的最有效手段,同时放疗是对恶性肿瘤最有效的非手术治疗。放射线和相当多的抗癌药物均可直接或间接作用于dna或dna代谢过程,从而导致dna损伤,其中,dna双链断裂(dna double strand break,dsb)对于癌细胞来讲是最致命的。dna损伤后,会引发受损dna修复等一系列细胞反应,而修复的结果就是提高癌细胞的存活,这也是肿瘤细胞对放化疗抵抗的机制之一。dna双链断裂如果不及时和完整修复,癌细胞则会因细胞凋亡或/和有丝分裂障碍导致细胞死亡。因此,只要抑制这些dna损伤的修复,就可以提高癌细胞对放化疗的敏感性,抑制细胞增殖。
4.在人等高等真核生物细胞中,dsb的修复主要是通过dna依赖性蛋白激酶(dna
‑
dependent protein kinase,dna
‑
pk)主导的dna非同源末端连接(nonhomologous end joining,nhej)进行,由此修复损伤的dna,保持细胞活性和基因组稳定性。nhej修复主要参与g1/s期dna损伤修复,并且不需要dna末端连接模板。nhej修复需要许多蛋白质和信号通路的共济协调。ku70/80亚单位的异质二聚体和催化亚单元dna依赖性蛋白激酶(dna
‑
pkcs),共同组成了有活性的dna
‑
pk酶复合物。
5.dna
‑
pkcs属于磷脂酰肌醇3激酶(pi3k)超家族成员,是一种丝氨酸/苏氨酸蛋白激酶;pi3k超家族还包括atm、atr、mtor和4个pi3k亚型。当dna
‑
pk与断裂的dna结合时,才能激活其激酶活性。ku的重要功能是与dna末端结合,招募dna
‑
pkcs,二者组成dna
‑
pk全酶并激活dna
‑
pkcs;活化的dna
‑
pkcs引导artemis蛋白(一种内切核酸酶)结合于受损位点,依靠其核酶活性进行dna断端处理以利于连接修复,然后xrcc4/dna
‑
连接酶iv复合物被活化的dna
‑
pkcs招募,最后由dna
‑
连接酶iv定位并连接断裂的dna双链末端,完成修复。xrcc4是与dna
‑
连接酶iv形成复合体的一种蛋白质,可以增加dna
‑
连接酶iv的活性。dna
‑
pkcs存在40个可自身磷酸化的氨基酸残基,最典型的自磷酸化位点发生在ser2056(por簇)和thr2609(abcde簇)。nhej被认为通过三个关键的步骤开展:识别dsb——ku70/80结合至不完全的dna末端,募集两分子的dna
‑
pkcs至dsb的相邻侧;进行dna加工以移除端点出的不可连接末端或其他损伤形式;最后连接dna末端。
6.由于肿瘤细胞具有较高基础水平的内源性复制压力(癌基因诱导的复制压力)和dna损伤,并且在肿瘤细胞中dna修复机制效率较低,因此肿瘤细胞对dna
‑
pk的敏感性更高。
7.目前,开发高效且选择性好的dna
‑
pk的抑制剂具有重要的临床意义,其可协同增强放疗和化疗效果,有效抑制肿瘤生长,同时可有效降低对正常细胞的损伤,减少副作用。
技术实现要素:
8.本发明要解决的技术问题是提供一种结构新颖的、对dna
‑
pk具有良好抑制作用的杂芳环类化合物。进一步的,该类化合物可用于增加受试者对于放疗和/或一种或多种抗癌剂的敏感性。更进一步的,该类化合物可与放疗和/或一种或多种抗癌剂联用用于预防和/或治疗良性肿瘤或癌症。
9.本发明的技术方案如下:
10.在一方面,本发明提供了如下通式(i)所示的化合物、其药学上可接受的盐或其异构体,
[0011][0012]
其中,
[0013]
x1、x2、x3分别独立地选自ch或n;
[0014]
x4选自cr2r3、nr4、o或s;
[0015]
x5选自cr5或n;
[0016]
y2、y3、y4、y5分别独立地选自ch或n;
[0017]
y选自s、nh或ch2;
[0018]
环a选自任选被1
‑
3个取代基取代的3
‑
8元环烷基或3
‑
8元杂环基,所述取代基选自卤素、羟基、氨基、硝基、氰基、c1‑6烷基、c1‑6烷基氨基、二(c1‑6烷基)氨基、卤代c1‑6烷基、羟基c1‑6烷基、氨基c1‑6烷基、c1‑6烷氧基、c1‑6烷硫基、c1‑6烷基羰基、卤代c1‑6烷氧基、卤代c1‑6烷硫基、羟基c1‑6烷氧基、羟基c1‑6烷硫基、氨基c1‑6烷氧基、氨基c1‑6烷硫基;
[0019]
r1、r2、r3、r4、r5分别独立地选自h、卤素、羟基、氨基、硝基、氰基、c1‑6烷基、c1‑6烷基氨基、二(c1‑6烷基)氨基、卤代c1‑6烷基、羟基c1‑6烷基、氨基c1‑6烷基、c1‑6烷氧基、c1‑6烷硫基、卤代c1‑6烷氧基、卤代c1‑6烷硫基、羟基c1‑6烷氧基、羟基c1‑6烷硫基、氨基c1‑6烷氧基、氨基c1‑6烷硫基。
[0020]
在某些实施方案中,环a选自任选被1
‑
2个取代基取代的3
‑
6元环烷基或3
‑
6元杂环基;所述取代基选自卤素、羟基、氨基、硝基、氰基、c1‑6烷基、c1‑6烷基氨基、二(c1‑6烷基)氨基、c1‑6烷基羰基、卤代c1‑6烷基、羟基c1‑6烷基、氨基c1‑6烷基、c1‑6烷氧基、c1‑6烷硫基、卤代c1‑6烷氧基或卤代c1‑6烷硫基。
[0021]
在某些实施方案中,环a选自3
‑
6元环烷基或3
‑
6元杂环基。
[0022]
在某些实施方案中,环a选自任选被1
‑
2个取代基取代的3
‑
6元饱和环烷基或3
‑
6元饱和杂环基;所述取代基选自卤素、羟基、氨基、硝基、氰基、c1‑6烷基、c1‑6烷基氨基、二(c1‑6烷基)氨基、c1‑6烷基羰基、卤代c1‑6烷基、羟基c1‑6烷基、氨基c1‑6烷基、c1‑6烷氧基、c1‑6烷硫基、卤代c1‑6烷氧基或卤代c1‑6烷硫基。
[0023]
在某些实施方案中,环a选自3
‑
6元饱和环烷基或3
‑
6元饱和杂环基。
[0024]
在某些实施方案中,环a选自任选被1
‑
2个取代基取代的环丙基、环丁基、环戊基、环己基、氧杂环丙基、氮杂环丙基、氧杂环丁基、氮杂环丁基、四氢呋喃基、四氢噻吩基、四氢吡咯烷基、四氢吡唑烷基、四氢咪唑烷基、四氢吡喃基、四氢噻喃基、哌啶基、哌嗪基、六氢嘧啶基、吗啉基;所述取代基选自卤素、羟基、氨基、硝基、氰基、甲基、乙基、丙基、异丙基、甲基氨基、二甲基氨基、甲基羰基、一氟甲基、二氟甲基、三氟甲基、羟基甲基、氨基甲基、甲氧基、乙氧基、丙氧基、异丙氧基、甲硫基、一氟甲氧基、二氟甲氧基或三氟甲氧基。
[0025]
在某些实施方案中,环a选自任选被1
‑
2个取代基取代的环戊基、环己基、氧杂环丁基、氮杂环丁基、四氢呋喃基、四氢噻吩基、四氢吡咯烷基、四氢吡喃基、四氢噻喃基、哌啶基、哌嗪基、吗啉基;所述取代基选自卤素、羟基、氨基、甲基、乙基、丙基、异丙基、甲基羰基、一氟甲基、二氟甲基、三氟甲基、羟基甲基、氨基甲基、甲氧基、乙氧基、丙氧基、异丙氧基、甲硫基、一氟甲氧基、二氟甲氧基或三氟甲氧基。
[0026]
在某些实施方案中,环a选自环戊基、环己基、氧杂环丁基、氮杂环丁基、四氢呋喃基、四氢吡咯烷基、四氢吡喃基、哌啶基、哌嗪基、吗啉基。
[0027]
在某些实施方案中,环a选自任选被1
‑
2个取代基取代的2个取代基取代的所述取代基选自卤素、羟基、氨基、甲基、乙基、丙基、异丙基、甲基羰基、一氟甲基、二氟甲基、三氟甲基、羟基甲基、氨基甲基、甲氧基、乙氧基、丙氧基、异丙氧基、甲硫基、一氟甲氧基、二氟甲氧基或三氟甲氧基。
[0028]
在某些实施方案中,r1、r2、r3、r4分别独立地选自h、卤素、羟基、氨基、硝基、氰基、c1‑6烷基、卤代c1‑6烷基、c1‑6烷氧基、c1‑6烷硫基、卤代c1‑6烷氧基或卤代c1‑6烷硫基;
[0029]
r5选自h、卤素、羟基、氨基、c1‑6烷基或卤代c1‑6烷基。
[0030]
在某些实施方案中,r1、r2、r3分别独立地选自氟、氯、溴、碘、羟基、氨基、甲基、乙基、丙基、异丙基、三氟甲基、甲氧基、乙氧基或三氟甲氧基。
[0031]
在某些实施方案中,r4选自h、甲基、乙基、丙基、异丙基或三氟甲基。
[0032]
在某些实施方案中,r5选自h、氟、氯、溴、碘、羟基、氨基、甲基、乙基或甲氧基。
[0033]
在某些实施方案中,y2、y3、y5分别为n,y4选自ch或n。
[0034]
在某些实施方案中,y2、y3、y5分别为n,y4为ch。
[0035]
在某些实施方案中,y为s。
[0036]
在某些实施方案中,x1、x2、x3分别独立地选自ch或n。
[0037]
在某些实施方案中,x1、x2分别独立地选自ch或n;x3为ch。
[0038]
在某些实施方案中,x1选自ch或n;x2为n;x3为ch。
[0039]
在某些实施方案中,x1、x2、x3分别独立地选自ch或n;
[0040]
x4选自cr2r3、nr4、o或s;
[0041]
x5为cr5或n;
[0042]
y2、y3、y5分别为n,y4为ch;
[0043]
y选自s或nh;
[0044]
环a选自任选被1
‑
2个取代基取代的环戊基、环己基、氧杂环丁基、氮杂环丁基、四氢呋喃基、四氢噻吩基、四氢吡咯烷基、四氢吡喃基、四氢噻喃基、哌啶基、哌嗪基、吗啉基;所述取代基选自氟、氯、溴、碘、羟基、氨基、甲基、乙基、丙基、异丙基、甲基羰基、三氟甲基、甲氧基、乙氧基、丙氧基、异丙氧基、甲硫基或三氟甲氧基;
[0045]
r1、r2、r3分别独立地选自氟、氯、溴、碘、羟基、氨基、甲基、乙基、丙基、异丙基、三氟甲基、甲氧基、乙氧基或三氟甲氧基;
[0046]
r4选自h、甲基、乙基、丙基、异丙基或三氟甲基;
[0047]
r5选自h、氟、氯、溴、碘、羟基、氨基、甲基、乙基或甲氧基。
[0048]
在某些实施方案中,x1、x2分别独立地选自ch或n;x3为ch;
[0049]
x4选自cr2r3、nr4、o或s;
[0050]
x5为cr5或n;
[0051]
y2、y3、y5分别为n,y4为ch;
[0052]
y选自s或nh;
[0053]
环a选自任选被1
‑
2个取代基取代的环己基、四氢呋喃基、四氢吡喃基、哌啶基;所述取代基选自氟、氯、溴、碘、羟基、甲基、乙基、异丙基、甲基羰基、三氟甲基、甲氧基、乙氧基或三氟甲氧基;
[0054]
r1、r2、r3分别独立地选自氟、氯、溴、碘、甲基、三氟甲基、甲氧基或三氟甲氧基;
[0055]
r4选自h、甲基、乙基、丙基、异丙基或三氟甲基;
[0056]
r5选自h、氟、氯、溴、碘、羟基、氨基、甲基、乙基或甲氧基。
[0057]
在某些实施方案中,通式(i)所述的化合物、其药学上可接受的盐或其异构体,其进一步具有如下通式(ii)所示的结构,
[0058][0059]
其中,r1、x1、x2、x4、r2、r3、r4、y、y3、y4、y5和环a如前文任一方案所述。
[0060]
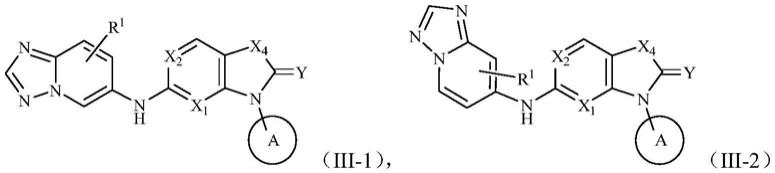
在某些实施方案中,通式(i)所述的化合物、其药学上可接受的盐或其异构体,其进一步具有如下通式(iii
‑
1)或通式(iii
‑
2)所示的结构,
[0061][0062]
其中,r1、x1、x2、x4、r2、r3、r4、y和环a如前文任一方案所述。
[0063]
本发明中各技术方案之间可以相互组合形成新的技术方案,所形成的新的技术方案同样包括在本发明的范围之内。
[0064]
在某些实施方案中,前述通式(i)所示的化合物、其药学上可接受的盐或其异构体,选自如下化合物:
[0065][0066][0067]
在另一个方面,本发明还提供一种药物制剂,含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,以及一种或多种药学上可接受的赋形剂,该药物制剂可为药学上可接受的任一剂型。药学上可接受的赋形剂是无毒性、与活性成分相容且其他方面生物学性质上适用于生物体的物质。特定赋形剂的选择将取决于用于治疗特定患者的给药方式或疾病类型和状态。
[0068]
在某些实施方案中,上述药物制剂可以以口服、肠胃外、直肠或经肺给药等方式施用于需要这种治疗的患者或受试者。用于口服给药时,所述药物组合物可制成口服制剂,例如可以制成常规的口服固体制剂,如片剂、胶囊剂、丸剂、颗粒剂等;也可制成口服液体制剂,如口服溶液剂、口服混悬剂、糖浆剂等。制成口服制剂时,可以加入适宜的填充剂、粘合剂、崩解剂、润滑剂等。用于肠胃外给药时,上述药物制剂也可制成注射剂,包括注射液、注射用无菌粉末与注射用浓溶液。制成注射剂时,可采用现有制药领域中的常规方法生产,配置注射剂时,可以不加入附加剂,也可以根据药物的性质加入适宜的附加剂。用于直肠给药时,所述药物组合物可制成栓剂等。用于经肺给药时,所述药物组合物可制成吸入制剂、气
雾剂、粉雾剂或喷雾剂等。
[0069]
在另一方面,本发明还涉及前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体在制备用于预防和/或治疗良性肿瘤或癌症等疾病的药物中的用途,所述癌症包括原位癌和转移的癌症。
[0070]
进一步的,本发明还涉及含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体的药物制剂在制备用于预防和/或治疗良性肿瘤或癌症等疾病的药物中的用途,所述癌症包括原位癌和转移的癌症。
[0071]
在另一方面,本发明还涉及前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体在制备用于预防和/或治疗良性肿瘤或癌症等疾病的药物中的用途,所述药物与放疗和/或一种或多种抗癌剂联用,所述癌症包括原位癌和转移的癌症。
[0072]
进一步的,本发明还涉及含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体的药物制剂在制备用于预防和/或治疗良性肿瘤或癌症等疾病的药物中的用途,所述药物可与放疗和/或一种或多种抗癌剂联用,所述癌症包括原位癌和转移的癌症。
[0073]
在另一方面,本发明还涉及前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体在制备用于使癌细胞对于抗癌剂和/或电离辐射敏感的药物中的用途。
[0074]
进一步的,本发明还涉及含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体的药物制剂在制备用于使癌细胞对于抗癌剂和/或电离辐射敏感的药物中的用途。
[0075]
所述的电离辐射是指患者在接受放疗过程中所接受的各种不同能量的射线的辐射。
[0076]
在另一个方面,本发明还提供一种药物组合物,其含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,以及一种或多种第二治疗活性剂,所述的第二治疗活性剂选自抗癌剂,包括有丝分裂抑制剂、烷化剂、抗代谢物、dna嵌合剂、抗肿瘤抗生素、生长因子抑制剂、信号传导抑制剂、细胞周期抑制剂、酶抑制剂、类维生素a受体调控剂、蛋白酶体抑制剂、拓扑异构酶抑制剂、生物应答调节剂、激素类药物、血管再生抑制剂、细胞生长抑制剂、靶向抗体、hmg
‑
coa还原酶抑制剂和异戊二烯基蛋白质转移酶抑制剂。
[0077]
在某些实施方案中,所述的第二治疗活性剂可以是减轻或降低本发明化合物在用于治疗受试者疾病时所产生的一种或多种副作用的药物,也可以是增强本发明化合物药效的药物。
[0078]
在某些实施方案中,所述药物组合物还包括一种或多种药学上可接受的赋形剂,所述赋形剂如前文所述。
[0079]
在另一方面,本发明还涉及含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体的药物组合物在制备用于预防和/或治疗良性肿瘤或癌症等疾病的药物中的用途,所述癌症包括原位癌和转移的癌症。
[0080]
在另一方面,本发明还涉及含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式
(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体的药物组合物在制备用于预防和/或治疗良性肿瘤或癌症等疾病的药物中的用途,所述药物可与放疗和/或一种或多种抗癌剂联用,所述癌症包括原位癌和转移的癌症。
[0081]
进一步,本发明还涉及含有前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体的药物组合物在制备用于使癌细胞对于抗癌剂和/或电离辐射敏感的药物中的用途。
[0082]
在另一方面,本发明还提供了一种治疗与dnapk过度活化相关的疾病的方法,该方法包括向有需要的患者施用有效量的前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,前述药物制剂或药物组合物;所述与dnapk过度活化相关的疾病选自良性肿瘤或癌症,所述的癌症包括原位癌和转移的癌症。
[0083]
进一步的,本发明还提供了一种治疗与dnapk过度活化相关的疾病的方法,该方法包括向接受放疗前/后的患者施用有效量的前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,前述药物制剂或药物组合物;所述与dnapk过度活化相关的疾病选自良性肿瘤或癌症,所述的癌症包括原位癌和转移的癌症。
[0084]
进一步的,本发明还提供了一种治疗与dnapk过度活化相关的疾病的方法,该方法包括向接受化疗前/后的患者施用有效量的前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,前述药物制剂或药物组合物;所述与dnapk过度活化相关的疾病选自良性肿瘤或癌症,所述的癌症包括原位癌和转移的癌症。
[0085]
在另一方面,本发明还提供了一种增强患者对于抗癌剂或放疗敏感性的方法,该方法包括向有需要的患者施用有效量的前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,前述药物制剂或药物组合物;所述的抗癌剂如下文所述。
[0086]
进一步,本发明还提供了一种增强患者对于抗癌剂或放疗敏感性的方法,该方法包括向接受放疗前/后的患者施用有效量的前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,前述药物制剂或药物组合物;所述的抗癌剂如下文所述。
[0087]
进一步,本发明还提供了一种增强患者对于抗癌剂或放疗敏感性的方法,该方法包括向接受化疗前/后的患者施用有效量的前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述化合物、其药学上可接受的盐或其异构体,前述药物制剂或药物组合物;所述的抗癌剂如下文所述。
[0088]
在另一方面,本发明还提供了一种试剂盒,包含:
[0089]
(a)有效量的一种或多种前述通式(i)、通式(ii)、通式(iii
‑
1)或通式(iii
‑
2)所述的化合物、其药学上可接受的盐或其异构体,
[0090]
和(b)有效量的一种或多种抗癌剂。
[0091]
本发明所述的“抗癌剂”是指对肿瘤具有一定治疗作用的药剂,包括但不限于有丝分裂抑制剂、烷化剂、抗代谢物、dna嵌合剂、抗肿瘤抗生素、生长因子抑制剂、信号传导抑制剂、细胞周期抑制剂、酶抑制剂、类维生素a受体调控剂、蛋白酶体抑制剂、拓扑异构酶抑制剂、生物应答调节剂、激素类药物、血管再生抑制剂、细胞生长抑制剂、靶向抗体、hmg
‑
coa还原酶抑制剂和异戊二烯基蛋白质转移酶抑制剂等;所述的肿瘤包括良性肿瘤和癌症。所述
的“有效量”是指能够预防、减轻、延缓、抑制或治愈受试者病症的药物剂量。给药剂量的大小与药物给药方式、药剂的药代动力学、疾病的严重程度、受试者的个性体征(性别、体重、身高、年龄)等相关。
[0092]
在本发明中,除非另有说明,否则本文中使用的科学和技术名词具有本领域技术人员通常理解的含义,然而为了更好地理解本发明,下面提供了部分术语的定义。当本发明所提供的术语的定义和解释与本领域技术人员所通常理解的含义不符的时候,以本发明所提供的术语的定义和解释为准。
[0093]
本发明所述的“卤素”是指氟原子、氯原子、溴原子或碘原子。
[0094]
本发明所述的“c1‑6烷基”表示直链或支链的含有1
‑
6个碳原子的烷基,包括例如“c1‑4烷基”、“c1‑3烷基”、“c1‑2烷基”、“c2‑6烷基”、“c2‑5烷基”、“c2‑4烷基”、“c2‑3烷基”、“c3‑6烷基”、“c3‑5烷基”、“c3‑4烷基”等,具体实例包括但不限于:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、2
‑
甲基丁基、新戊基、1
‑
乙基丙基、正己基、异己基、3
‑
甲基戊基、2
‑
甲基戊基、1
‑
甲基戊基、3,3
‑
二甲基丁基、2,2
‑
二甲基丁基、1,1
‑
二甲基丁基、1,2
‑
二甲基丁基、1,3
‑
二甲基丁基、2,3
‑
二甲基丁基、2
‑
乙基丁基、1,2
‑
二甲基丙基等。本发明所述的“c1‑4烷基”指c1‑6烷基中的含有1
‑
4个碳原子的具体实例。
[0095]
本发明所述的“c1‑6烷氧基”是指“c1‑6烷基
‑
o
‑”
,所述的“c1‑6烷基”如前文所定义。本发明所述的“c1‑4烷氧基”是指“c1‑4烷基
‑
o
‑”
,所述的“c1‑4烷基”如前文所定义。
[0096]
本发明所述的“c1‑6烷硫基”是指“c1‑6烷基
‑
s
‑”
,所述的“c1‑6烷基”如前文所定义。本发明所述的“c1‑4烷硫基”是指“c1‑4烷基
‑
s
‑”
,所述的“c1‑4烷基”如前文所定义。
[0097]
本发明所述的“羟基c1‑6烷基、氨基c1‑6烷基、卤代c1‑6烷基”是指c1‑6烷基中的一个或多个氢分别被一个或多个羟基、氨基或卤素所取代。c1‑6烷基如前文所定义
[0098]
本发明所述“羟基c1‑6烷氧基、氨基c1‑6烷氧基、卤代c1‑6烷氧基”是指“c1‑6烷氧基”中的一个或多个氢被一个或多个羟基、氨基或卤素所取代。
[0099]
本发明所述“羟基c1‑6烷硫基、氨基c1‑6烷硫基、卤代c1‑6烷硫基”是指“c1‑6烷硫基”中的一个或多个氢被一个或多个羟基、氨基或卤素所取代。
[0100]
本发明所述的“c1‑6烷基氨基、二(c1‑6烷基)氨基”分别是指c1‑6烷基
‑
nh
‑
、
[0101]
本发明所述的“c1‑6烷基羰基”是指c1‑6烷基
‑
c(o)
‑
。
[0102]
本发明所述的“3
‑
8元环烷基”是指含有3
‑
8个环原子的饱和或部分饱和的且不具有芳香性的单环环状基团,本发明所述的“3
‑
8元环烷基”包括“3
‑
8元饱和环烷基”和“3
‑
8元部分饱和环烷基”,例如是“3
‑
6元环烷基”、“3
‑
6元饱和环烷基”、“5
‑
6元环烷基”、“5
‑
6元饱和环烷基”等。其实例包括但不限于:环丙基、环丁基、环戊基、环己基或环己烯基等。
[0103]
本发明所述的“3
‑
8元杂环基”是指至少含有一个(例如,含有1个、2个、3个、4个或5个)杂原子的且环原子数为3
‑
8个的饱和或部分饱和的且不具有芳香性的单环环状基团,所述杂原子为氮原子、氧原子和/或硫原子,任选地,环状结构中的环原子(例如碳原子、氮原子或硫原子)可以被氧代。本发明所述的“3
‑
8元杂环基”包括“3
‑
8元饱和杂环基”和“3
‑
8元部分饱和杂环基”。所述“3
‑
8元杂环基”例如是“3
‑
6元杂环基”、“3
‑
6元饱和杂环基”、“3
‑
5元杂环基”、“3
‑
5元饱和杂环基”、“5
‑
6元杂环基”、“5
‑
6元饱和杂环基”等。其具体实例包括但
不仅限于:氮杂环丙烷基、2h
‑
氮杂环丙烷基、二氮杂环丙烷基、3h
‑
二氮杂环丙烯基、氮杂环丁烷基、氧杂环丙基、氧杂环丁烷基、1,4
‑
二氧杂环己烷基、1,3
‑
二氧杂环己烷基、1,3
‑
二氧杂环戊烷基、1,4
‑
二氧杂环己二烯基、四氢呋喃基、二氢吡咯基、四氢吡咯烷基、四氢吡唑烷基、四氢咪唑烷基、4,5
‑
二氢咪唑基、吡唑烷基、4,5
‑
二氢吡唑基、2,5
‑
二氢噻吩基、四氢噻吩基、4,5
‑
二氢噻唑基、噻唑烷基、四氢吡喃基、四氢噻喃基、哌啶基、四氢吡啶基、哌啶酮基、四氢吡啶酮基、二氢哌啶酮基、哌嗪基、六氢嘧啶基、吗啉基等。
[0104]
本发明所述“任选被取代基取代”是指被取代的基团上的一个或多个氢原子被一个或多个取代基“取代”或者“不取代”的两种情形。
[0105]
本发明所述的“化疗”是化学药物治疗的简称,主要通过使用化学治疗药物杀灭癌细胞达到治疗的目的。
[0106]
本发明所述的“放疗”是指一种肿瘤治疗方法,即肿瘤放射治疗,主要利用放射线进行肿瘤局部治疗,所述的“放射线”包括放射性同位素产生的α、β、γ射线和各类x射线治疗机或加速器产生的x射线、电子线、质子束及其他粒子束等。
[0107]
本发明所述的“药学上可接受的盐”是指化合物中存在的酸性官能团(例如
‑
cooh、
‑
oh、
‑
so3h等)与适当的无机或者有机阳离子(碱)形成的盐,包括与碱金属或碱土金属形成的盐、铵盐、与含氮有机碱形成的盐;以及化合物中存在的碱性官能团(例如
‑
nh2等)与适当的无机或者有机阴离子(酸)形成的盐,包括与无机酸或有机酸(例如羧酸等)形成的盐。
[0108]
本发明所述的“异构体”是指本发明化合物含有一个或多个不对称中心,因而可作为外消旋体和外消旋混合物、单一对映异构体、非对映异构体混合物和单一非对映异构体。本发明化合物可以有不对称中心,这类不对称中心各自独立地产生两个光学异构体。本发明的范围包括所有可能的光学异构体和它们的混合物。本发明所述的化合物若含有烯烃双键,除非特别说明,包括顺式异构体和反式异构体。本发明所述的化合物可以以互变异构体(官能团异构体的一种)形式存在,其通过一个或多个双键位移而具有不同的氢的连接点,例如,酮和它的烯醇形式是酮
‑
烯醇互变异构体。本发明化合物含有螺环结构,受环的立体空间结构的影响,环上的取代基可存在于环两侧从而形成相对的顺式(cis)和反式(trans)异构体。各互变异构体及其混合物都包括在本发明的范围中。所有化合物的对映异构体、非对映异构体、外消旋体、内消旋体、顺反异构体、互变异构体、几何异构体、差向异构体及其混合物等,均包括在本发明范围中。
[0109]
本发明化合物可通过对映体特异性合成或从对映异构体混合物拆分以得到个别对映异构体的形式制备。常规拆分技术包括使用各种众所周知的色谱方法拆分起始物质或最终产物的对映异构体的混合物。
[0110]
当公开的化合物的立体化学通过结构命名或描绘时,命名或描绘的立体异构体相对于其他立体异构体为至少60%重量、70%重量、80%重量、90%重量、99%重量或99.9%重量纯。当单一异构体通过结构命名或描绘时,所描绘或命名的对映异构体为至少60%重量、70%重量、80%重量、90%重量、99%重量或99.9%重量纯。光学纯度重量%为对映异构体的重量与对映异构体重量加上其光学异构体的重量比率。
[0111]
发明的有益效果
[0112]
1、本发明化合物、其药学上可接受的盐或其异构体具有优异的dna
‑
pk抑制作用,
其在生物体内具有良好的药代动力学性质,作用持久,生物利用度高,能够增强癌细胞对放疗和/或一种或多种抗癌剂的敏感性。
[0113]
2、本发明化合物、其药学上可接受的盐或其异构体酶学选择性高、对良性肿瘤和癌症具有较好的治疗作用,且肝微粒体稳定性高。
[0114]
3、本发明化合物制备工艺简单,药品纯度高,质量稳定,易于进行大规模工业生产。
具体实施方案
[0115]
下面将结合具体实施方式对本发明技术方案进行描述,对本发明的上述内容作进一步的详细说明,但不应将此理解为本发明上述主题的范围仅限于以下实施例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
[0116]
缩写:
[0117]
dmfdma:n,n
‑
二甲基甲酰胺二甲基缩醛;dppa:叠氮磷酸二苯酯;劳森试剂:2,4
‑
双(对甲氧苯基)
‑
1,3
‑
二硫杂
‑
2,4
‑
二磷杂环丁烷
‑
2,4硫化物;diea:n,n
‑
二异丙基乙胺;dcm:二氯甲烷;meoh:甲醇;brettphos pd g3:甲烷磺酸(2
‑
二环己基膦)
‑
3,6
‑
二甲氧基
‑
2',4',6'
‑
三异丙基
‑
1,1'
‑
联苯)(2'
‑
氨基
‑
1,1'
‑
联苯
‑2‑
基)钯(ii)。
[0118]
制备例一:7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
胺的制备
[0119]
1、(e)
‑
n,n
‑
二甲基
‑
n
’‑
(4
‑
甲基
‑5‑
硝基吡啶
‑2‑
基)甲脒的制备
[0120][0121]
将4
‑
甲基
‑5‑
硝基吡啶
‑2‑
胺(20g,130.6mmol),甲苯(400ml),加入n,n
‑
二甲基甲酰胺二甲基缩醛(47g,394.3mmol),加热至110℃,反应3小时,旋干,加入正庚烷(300ml)浆洗,过滤,固体干燥得目标产物(26g,产率:95.6%)。
[0122]
2、(e)
‑
n
‑
羟基
‑
n
’‑
(4
‑
甲基
‑5‑
硝基吡啶
‑2‑
基)甲脒的制备
[0123][0124]
(e)
‑
n,n
‑
二甲基
‑
n
’‑
(4
‑
甲基
‑5‑
硝基吡啶
‑2‑
基)甲脒(16.0g,76.8mmol),甲醇(200ml),加入盐酸羟胺(10.7g,153.9mmol),加热至65℃,3小时,冷却,加入水(300ml),乙酸乙酯(300ml),分液,有机相用无水硫酸钠干燥,过滤,旋干,固体用甲基叔丁基醚(100ml)浆洗,固体干燥得目标产物(9.2g,产率:61.3%)。
[0125]
3、7
‑
甲基
‑6‑
硝基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶的制备
[0126][0127]
(e)
‑
n
‑
羟基
‑
n
’‑
(4
‑
甲基
‑5‑
硝基吡啶
‑2‑
基)甲脒(10.8g,55.0mmol),四氢呋喃(110ml),加入三氟乙酸酐(13.9g,66.2mmol),10℃,反应12小时,lc
‑
ms检测反应完成,将溶剂旋干,固体中加入混合溶剂(100ml,乙酸乙酯:石油醚=1:4),过滤,固体真空干燥得目标产物(5.6g,产率:57.1%)。
[0128]
4、7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
胺的制备
[0129][0130]
将7
‑
甲基
‑6‑
硝基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶(4.4g,24.7mmol)溶于乙醇(50ml)中,加入钯碳(528mg),甲酸铵(7.8g,124.0mmol),加热至70℃,反应4小时,抽滤,滤液旋干得产物(3.6g,产率:98.3%)。
[0131]
制备例二:2
‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
酮的制备
[0132]
1、2
‑
氯
‑4‑
((四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)嘧啶
‑5‑
羧酸乙酯的制备
[0133][0134]
将2,4
‑
二氯嘧啶
‑5‑
羧酸乙酯(40.0g,181mmol)溶于乙腈(1000ml)中,加入四氢
‑
2h
‑
吡喃
‑4‑
胺盐酸盐(24.9g,181mmol),碳酸钾(62.5g,452mmol),20℃,反应16小时,lc
‑
ms检测反应完成,抽滤,滤液浓缩得目标产物(45g,产率:87.0%)。
[0135]
2、2
‑
氯
‑4‑
((四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)嘧啶
‑5‑
羧酸的制备
[0136][0137]
将2
‑
氯
‑4‑
((四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)嘧啶
‑5‑
羧酸乙酯(15.0g,52.4mmol)溶于四氢呋喃(150.0ml)和水(150.0ml)体系中,加入氢氧化锂(4.8g,114.3mmol),25℃,反应1小时,浓缩,水相调ph=3析出白固体,过滤,固体干燥得目标产物(11.5g,产率:85.2%)。
[0138]
3、2
‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
酮的制备
[0139][0140]
将2
‑
氯
‑4‑
((四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)嘧啶
‑5‑
羧酸(10.0g,38.8mmol)溶于n,n
‑
二甲基乙酰胺(60.0ml)中,加入三乙胺(4.0g,39.6mmol)和叠氮磷酸二苯酯(11.0g,40.0mmol),体系在25℃反应1小时后,升至110℃继续反应16小时,降至25℃后倒入冰水中,析出淡黄色固体,过滤旋干得粗品(7.1g,产率:71.7%)。
[0141]
制备例三:2
‑
氯
‑7‑
甲基
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
酮的制备
[0142][0143]
将2
‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
酮(7.4g,29.0mmol)溶于n,n
‑
二甲基甲酰胺(100.0ml)中,体系降至0℃后加入60%nah(1.5g,37.5mmol)反应30分钟后加入ch3i(5.0g,35.2mmol),在0℃下继续反应2小时后,加入水(150.0ml)和乙酸乙酯(220.0ml),有机相经浓缩旋干得粗品(6.9g,产率:88.5%)。
[0144]
实施例一:7
‑
甲基
‑2‑
((7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
基)氨基)
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
硫酮(化合物1)的制备
[0145]
1、7
‑
甲基
‑2‑
((7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
基)氨基)
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
酮的制备
[0146][0147]
将2
‑
氯
‑7‑
甲基
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
酮(3.3g,12.3mmol)溶于二氧六环(50.0ml)中,加入7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
胺(1.8g,12.2mmol),2
‑
(二环己基膦)3,6
‑
二甲氧基
‑2′
,4
′
,6
′‑
三异丙基
‑
1,1
′‑
联苯(760.0mg,0.784mmol)和碳酸铯(7.8g,23.9mmol)体系在氮气环境下100℃反应2.0小时后进行浓缩,用甲醇(50.0ml)洗涤。固体物进行柱层析(dcm:meoh=20:1)得产物(3.4g,产率:73.9%)。
[0148]
分子式:c
18
h
20
n8o
2 分子量:380.4 lc
‑
ms(m/e):381.2(m h
)
[0149]1hnmr(400mhz,dmso):δ9.10(s,1h),8.66(s,1h),8.35(s,1h),8.06(s,1h),7.69(s,1h),4.41
‑
4.37(m,1h),3.96
‑
3.92(m,2h),3.42
‑
3.39(m,2h),3.28(s,3h),2.53
‑
2.50(m,1h),2.48
‑
2.35(m,1h),2.37(s,3h),1.67
‑
1.64(m,2h)
[0150]
2、7
‑
甲基
‑2‑
((7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
基)氨基)
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
硫酮的制备
[0151][0152]
将7
‑
甲基
‑2‑
((7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
基)氨基)
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑
酮(250.0mg,0.656mmol),劳森试剂(1400.0mg,3.456mmol)溶于二氧六环(10.0ml)中,体系在155℃下微波反应4.0小时,浓缩后残余物进行柱层析(二氯甲烷:甲醇=20:1)得产物(35.0mg,收率为13.5%).
[0153]
分子式:c
18
h
20
n8os 分子量:396.5 lc
‑
ms(m/e):397.2(m h
)
[0154]1h
‑
nmr(400mhz,dmso)δ:9.06(s,1h),8.47(s,1h),8.40(s,1h),7.75(s,1h),5.25(s,1h),4.0
‑
13.91(m,2h),3.7(s,3h),3.52
‑
3.42(m,2h),2.72
‑
2.63(m,2h),2.48(s3h),1.77
‑
1.68(m,2h).
[0155]
实施例二:8
‑
亚氨基
‑7‑
甲基
‑
n
‑
(7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
基)
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
8,9
‑
二氢
‑
7h
‑
嘌呤
‑2‑
胺(化合物2)的制备
[0156]
1、2,8
‑
二氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
9h
‑
嘌呤的制备
[0157][0158]
将2
‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
酮(6.7g,26.3mmol)溶于pocl3(60.0ml)中,向体系中加入diea(5.9g,45.8mmol),然后升温至110℃反应11.0小时后,经浓缩旋干后用饱和nahco3水溶液调ph为7,再浓缩后对粗品进行柱层析(dcm:meoh=10:1)纯化得产物(3.82g,产率:53.2%)。
[0159]
2、2
‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
9h
‑
嘌呤
‑8‑
胺的制备
[0160][0161]
将2,8
‑
二氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
9h
‑
嘌呤(3.2g,11.7mmol)溶于胺甲醇溶液中(7m,50.0ml),体系在100℃反应3.0小时后进行浓缩,经柱层析(meoh:dcm=1:10)纯化得产物(1.5g,产率:50.5%)。
[0162]
3、2
‑
氯
‑7‑
甲基
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
亚胺的制备
[0163][0164]
将2
‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
9h
‑
嘌呤
‑8‑
胺(530mg,2.1mmol)溶于乙醇
(15.0ml)中,然后加入ch3i(2.0g,14.1mmol)体系在130℃下微波反应2.0小时后进行浓缩,经柱层析(meoh:dcm=1:10)纯化得粗品产物(120.0mg,产率:21.3%)。
[0165]
4、8
‑
亚氨基
‑7‑
甲基
‑
n
‑
(7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
基)
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
8,9
‑
二氢
‑
7h
‑
嘌呤
‑2‑
胺的制备
[0166][0167]
将2
‑
氯
‑7‑
甲基
‑9‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑
7,9
‑
二氢
‑
8h
‑
嘌呤
‑8‑
亚胺(89.9mg,0.34mmol),7
‑
甲基
‑
[1,2,4]三唑并[1,5
‑
a]吡啶
‑6‑
胺(50.0mg,0.34mmol),碳酸铯(220.0mg,0.67mmol)和brettphos pd g3(25.0mg,0.028mmol)溶于二氧六环(5.0ml)中,然后体系在n2环境下100℃反应3.0小时后进行浓缩,经柱层析(meoh:dcm=1:10)纯化得产物(7.5mg,产率:5.8%)。
[0168]
分子式:c
18
h
21
n9o 分子量:379.4 lc
‑
ms(m/e):380.0(m h
)
[0169]1hnmr(400mhz,cdcl3):δ:9.84(s,1h),8.25(s,1h),7.68(s,1h),7.56(s,1h),7.26(s,1h),6.59(s,1h),4.67
‑
4.45(m,1h),4.17
‑
4.15(m,2h),3.58
‑
3.49(m,2h),3.31(s,3h),2.91
‑
2.75(m,2h),2.53(s,3h),1.87
‑
1.76(m,2h).
[0170]
使用与上述实例相同或相似的方法制备了以下表格所示的化合物:
[0171][0172]
实验方案
[0173]
以下提供本发明部分化合物的示例性实验方案,以显示本发明化合物有利活性和有益技术效果。但是应当理解,下述实验方案仅仅是对本发明内容的示例,而不是对本发明范围的限制。
[0174]
实验例1本发明化合物的体外细胞学活性
[0175]
缩写
[0176]
edta:乙二胺四乙酸
[0177]
dmso:二甲基亚砜
[0178]
tris:三羟甲基氨基甲烷
[0179]
brij
‑
35:月桂醇聚氧乙烯醚
[0180]
dtt:二硫苏糖醇
[0181]
供试品:本发明化合物,其结构式、制备方法见实施例。
[0182]
实验试剂:
[0183]
名称品牌adp
‑
glo kinase assaypromegedna
‑
pkpromege
[0184]
实验方法:
[0185]
1.配制1倍的激酶缓冲液
[0186]
1)1倍激酶缓冲液
[0187]
40mm tris,ph 7.5
[0188]
0.0055%brij
‑
35
[0189]
20mm mgcl2[0190]
0.05mm dtt
[0191]
2.化合物配制
[0192]
1)化合物的检测起始浓度为1μm,配制成100倍浓度,即100μm。取2μl 10mm化合物,加入198μl 100%dmso,配制成100μm化合物溶液。在96孔板上第二个孔中加入100μl的100倍的化合物,其他孔加入60μl的100%dmso。从第二孔中取30μl化合物加入第三孔中,依次往下做3倍稀释,共稀释10个浓度。
[0193]
2)转移100μl 100%dmso和阳性对照wortmannin的最高浓度(400nm)分别到两个空的孔中作为max孔和min孔。
[0194]
3)使用echo转移50nl化合物到384孔板中。
[0195]
3.配制2x激酶溶液
[0196]
1)使用1倍激酶缓冲液配制2倍dna
‑
pk激酶溶液。
[0197]
2)转移2.5μl 2倍酶溶液到384孔板反应孔中。
[0198]
3)振荡,混匀,室温下静置。
[0199]
4.配制2x底物溶液
[0200]
1)使用1倍激酶缓冲液配制2倍底物溶液。
[0201]
2)转移2.5μl 2倍底物溶液到384孔板反应孔中起始反应。
[0202]
3)振荡,混匀。
[0203]
5.激酶反应和终止
[0204]
1)将384孔板盖上盖子,于28℃下孵育3小时。
[0205]
2)转移5μl adp
‑
glo reagent,于28℃下孵育2小时。
[0206]
6.反应结果的检测
[0207]
1)转移10μl激酶检测试剂到384孔板反应孔中终止反应。
[0208]
2)室温下静止30分钟。
[0209]
7.数据读取
[0210]
在envision读取样品数值。
[0211]
8.抑制率计算
[0212]
将数据转化为抑制率数据后进行曲线拟合。
[0213]
抑制百分数=(max
‑
conversion)/(max
‑
min)*100.其中max是指dmso对照的转化率,min是指无酶活对照的转化率,conversion是指测试化合物各浓度下的转化率。
[0214]
实验结果:
[0215]
表1本发明化合物体外酶学活性数据
[0216][0217]
实验结论:
[0218]
结果表明,本发明化合物对dna
‑
pk激酶活性有较好的抑制作用。
[0219]
实验例2本发明化合物在不同种属中的肝微粒体代谢稳定性实验
[0220]
供试品:本发明化合物1,自制,其化学名称和制备方法见化合物的制备实施例。
[0221]
实验材料:
[0222]
食蟹猴混合肝微粒体购自瑞德肝脏疾病研究中心(上海有限公司),批号为:zxbz,肝微粒体蛋白浓度为20mg
·
ml
‑1。
[0223]
sd大鼠、cd
‑
1小鼠的混合肝微粒体均购自xeno tech,批号分别为:1610290(sd大鼠)、1710069(cd
‑
1小鼠)。肝微粒体蛋白浓度均为20mg
·
ml
‑1。
[0224]
人混合肝微粒体购自于corning公司,货号为452117,批号为38294,肝微粒体蛋白浓度为20mg
·
ml
‑1。
[0225]
实验启动因子β
‑
nadph购于solarbio公司;ph 7.4的磷酸盐缓冲液(pbs)由本实验室自制。
[0226]
供试品溶液制备:
[0227]
精密称取供试品粉末适量,加入适量的二甲基亚砜(dmso)溶解到1mm,再用甲醇稀释20倍到50μm的工作液。
[0228]
实验方法:
[0229]
表2.肝微粒体代谢稳定性实验温孵体系组成
[0230][0231]
实验操作步骤:
[0232]
(1)按照上面表2“实验温孵体系构成”的比例,每个化合物取100mm pbs 5.85ml,20mm mgcl2溶液0.585ml及h2o 3.57ml,制备温孵体系混合溶液1(不含微粒体、供试品和β
‑
nadph)。该实验同时进行实验孵育体系的阳对药维拉帕米以证明肝微粒体酶活性正常。
[0233]
(2)从
‑
80℃冰箱中取出肝微粒体(20mg蛋白/ml),置于37℃水浴恒温振荡器上预温孵3min。
[0234]
(3)每个化合物每个种属取1.9ml温孵体系混合溶液1,加入56μl不同种属的微粒体,制备温孵体系混合溶液2(不含供试品和β
‑
nadph)。
[0235]
(4)样品组(含微粒体和β
‑
nadph):取616μl温孵体系混合溶液2,加入14μl浓度为50μm的供试品工作溶液,加入70μl 10mm的β
‑
nadph工作溶液。混匀,复样。取样时间点为0min,5min,10min,20min,30min,60min。该样品组用于评价化合物经由β
‑
nadph介导的代谢稳定性。
[0236]
(5)对照组(含微粒体,不含β
‑
nadph,以水代替β
‑
nadph):取264μl温孵体系混合溶液2,加入6μl浓度为50μm的供试品工作溶液,加入30μl水。混匀,复样。取样时间点为0min和60min。该阴性对照组用于评价化合物在肝微粒体孵育体系中是否存在非β
‑
nadph介导的代谢。
[0237]
(6)于各个预定时间点从孵育样品管中取样50μl,加入至终止样品管(内含300μl冷的终止剂,含内标甲苯磺丁脲50ng/ml的乙腈溶液),涡旋,终止反应。
[0238]
(7)涡旋10min后,离心5min(12000rpm)。
[0239]
(8)取上清液100μl,加入100μl水,涡旋混匀,lc
‑
ms/ms进样分析。
[0240]
数据分析:
[0241]
通过下面公式中供试品与内标峰面积的比值转化成剩余量百分比。
[0242][0243]
实验结果:
[0244]
表3本发明化合物肝微粒体稳定性结果
[0245][0246]
实验结论:
[0247]
本发明化合物在人、猴、大鼠及小鼠肝微粒体中具有良好的稳定性。
[0248]
实验例3本发明化合物的cd1小鼠体内药代动力学实验
[0249]
缩写
[0250]
hpc:羟丙基纤维素
[0251]
dma:二甲基乙酰胺供试品:本发明化合物1,自制,其化学名称和制备方法见化合物的制备实施例。受试动物:cd1小鼠,雌性,购自北京维通利华实验动物技术有限公司,体重24
‑
29g,共12只。
[0252]
供试品溶液制备:
[0253]
空白溶媒(1)的配制方法:称取28g hp
‑
β
‑
cd,加入适量注射用水溶解,再用注射用水定容到100ml,涡旋混匀,即得28%hp
‑
β
‑
cd。
[0254]
空白溶媒(2)的配制方法:称取20g hpc,缓慢加入500ml搅拌着的纯化水中,再加入1ml吐温80,搅拌至澄清透明,定容到1000ml,搅拌均匀即得2%hpc 0.1%吐温80。
[0255]
iv(静脉推注)给药:
[0256]
取化合物1(2.57mg),加入dma(491μl),振摇溶解,然后加入peg400(982μl),涡旋混匀,最后加入空白溶媒(1)(3.44ml),涡旋混匀,50℃保温20min,即得0.5mg/ml的澄清溶液,作为化合物1的iv给药溶液。
[0257]
po(灌胃)给药:
[0258]
称取化合物1(3.83mg),置于组织研磨器中,加入空白溶媒(2)3.66ml,以1200转/分钟研磨均匀,即得浓度为1mg/ml的混悬药液,作为化合物1的po给药药液。
[0259]
实验方法
[0260]
iv给药剂量为2.5mg/kg,给药浓度为0.5mg/ml,给药体积为5ml/kg。
[0261]
po给药剂量为10mg/kg,给药浓度为1mg/ml,给药体积为10ml/kg。
[0262]
采血时间点:给药后0.083、0.25、0.5、1、2、4、6、8、24h,具体按下表所示方式采血。
[0263][0264]
每个时间点通过眼内眦采集全血约50μl,放置到含有edta
‑
k2抗凝剂的抗凝管中,在4℃条件下8000转/分钟离心6分钟得到血浆样品,血浆于
‑
80℃冰箱冻存,待分析。
[0265]
血浆样品分析
[0266]
采用蛋白沉淀法:取血浆样品20μl,加入内标(含甲苯磺丁脲50ng/ml的乙腈溶液)200μl,涡旋10min后,然后4000转/分钟离心20分钟,取上清液100μl,再加入100μl水,涡旋混匀3min后,lc
‑
ms/ms分析。
[0267]
实验结果
[0268]
表4 cd1小鼠pk评价结果
[0269][0270]
auc0‑
t
代表药时曲线下面积0
→
t;cl代表清除率;v
ss
表示稳态表观分布容积;t
1/2
代表末端消除半衰期;t
max
代表达峰时间;c
max
代表达峰浓度;f%代表绝对生物利用度。
[0271]
实验结论
[0272]
由表4的数据可知,本发明化合物具有良好的药代动力学性质,且具有较高的暴露量和生物利用度。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

