一种wnt通路抑制剂
技术领域
1.本发明涉及一种wnt通路抑制剂及其制备方法和应用。
背景技术:
2.wnt信号通路在多细胞生物体轴分化、组织器官发生、肿瘤形成等生命过程中具有重要作用。wnt基因编码表达的蛋白是一类分泌型糖蛋白,由19个成员组成,通过与细胞膜上frizzled(fzd)家族蛋白及低密度脂蛋白受体相关蛋白(ldl receptor related protein,lrp)受体结合,激活典型wnt/β-catenin通路、平面极通路、wnt/ca
2
通路等多种细胞内信号途径,调控着包括增殖、分化、死亡、迁移、极化等多种细胞功能(nusse roel,varmus harold e.(1992).wnt genes.cell,69(7),1073-1087.)。
3.经研究报道发现,神经性疾病、炎症纤维化疾病、代谢性疾病以及多种类型癌症的发病机制中都有涉及到典型wnt/β-catenin信号通路β-catenin-tcf/lcf转录复合物的激活的失调(kahn,m.(2014).can we safely target the wnt pathway?nature reviews.nat.rev.drug.discovery,13(7),513-532.)。在癌症研究领域中,wnt信号参与肿瘤早期证据来源于小鼠乳腺癌中分离得到因病毒插入而激活的癌基因int1;近10%的结直肠癌、头颈癌、肺癌、卵巢癌、黑色素瘤患者癌症发生与wnt信号调控元件r-spondin家族和rnf43/znrf3的功能突变诱导相关(b madan,z ke,n harmston,et al.(2015).wnt addiction of genetically defined cancers reversed by porcn inhibition.oncogene,1-11.)。
技术实现要素:
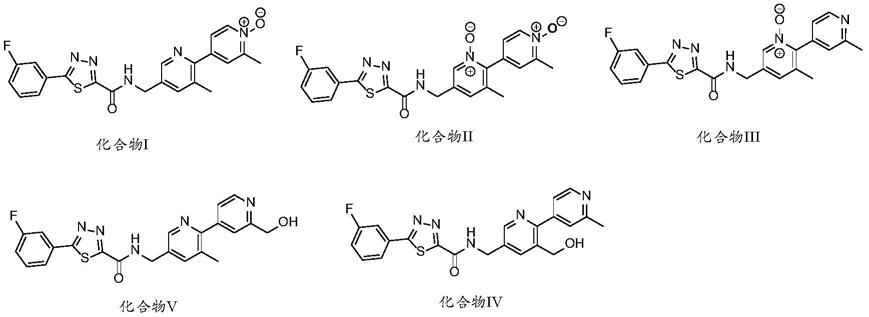
4.本发明一方面提供了如下式所示的化合物:
[0005][0006]
本发明第二方面提供了这些化合物的制备方法。
[0007]
本发明第三方面提供了这些化合物在制备作为wnt通路抑制剂的药物中的应用。
具体实施方式
[0008]
以下是本发明的具体实施例,对本发明的技术方案做进一步的描述,但是本发明
的保护范围并不限于这些实施例。凡是不背离本发明构思的改变或等同替代均包括在本发明的保护范围之内。
[0009]
实施例1化合物i的制备
[0010][0011]
单口瓶中将rex-p25-9(1g,2.38mmol)溶于二氯甲烷(10ml)中,将反应液用冰水浴降温至0℃,缓慢加入m-cpba(0.7eq,287.97mg,1.67mmol)。体系在冰水浴中反应20min,爬小板(dcm:meoh=15:1)显示rex-p25-9原料点基本消失。将反应液倒入30ml的冰水中,用二氯甲烷(10ml)萃取三次,饱和食盐水洗涤一次,无水硫酸镁干燥,过滤除去固体,旋转蒸发除去溶液,得到白色不溶物固体。将白色不溶物固体尽量多的溶解于二氯甲烷中,用大板提纯,得到化合物i(510mg)。ms(esi)m/z=436.18[m h]
。1h nmr(400mhz,cdcl3)δ8.56(d,j=1.7hz,1h),8.33(d,j=6.7hz,1h),7.95(t,j=5.9hz,1h),7.79
–
7.73(m,2h),7.67(d,j=1.5hz,1h),7.56
–
7.46(m,2h),7.37
–
7.23(m,1h),4.74(d,j=6.2hz,2h),2.58(s,3h),2.43(s,3h)。
[0012]
实施例2化合物v的制备
[0013][0014]
化合物2的合成:
[0015]
向三口瓶中加入化合物1(rex-p25-9,20g)、二氯甲烷(200ml)、无水甲醇(200ml)、m-cpba(8.24g),搅拌溶清,保温20~25℃反应17h;取样检测原料未反应完,补加m-cpba(8.24g),保温20~25℃反应23h;将反应液降温至<10℃,有白色固体析出,抽滤,滤饼减压45℃真空干燥后得到白色固体2(18.5g)。
[0016]
化合物3的合成:
[0017]
向三口瓶中加入化合物2(18.5g),乙酸酐(185ml),搅拌升温至60~70℃,保温反应16h;取样检测,有目标分子,将反应液降温至20~25℃,有固体析出,过滤,滤饼真空干燥后得到6.9g粗品,柱层析(pe
→
pe:ea
→
dcm:meoh=10:1),合并层析液浓缩至干得到淡黄色固体化合物3(2.5g)。
[0018]
化合物v的合成:
[0019]
向三口瓶中加入化合物3(2.5g),thf(12.5ml),纯化水(12.5ml),氢氧化钠水溶液
(2.48ml,含251mg氢氧化钠),搅拌升温至75~80℃;保温反应50min,取样检测,原料反应完全;将反应液冷却至室温,用稀盐酸调ph至7~8,过滤,滤饼用少量纯化水洗涤,真空干燥后得到1.1g粗品;粗品送制备分离,得到化合物v(759mg)。ms(esi)m/z=436.8[m h]
。1h nmr(400mhz,dmso-d6)δ10.03-10.00(t,1h),8.57-8354(m,2h),7.94-7.75(d,2h),7.74(s,1h),7.67-7.62(m,2h),7.53-7.48(m,1h),7.43-7.42(m,1h),5.50-5.47(t,1h),4.63-4.57(m,4h),2.35(s,3h)。
[0020]
实施例3化合物iii的制备
[0021][0022]
化合物2的合成:
[0023]
向100ml单口瓶中加入化合物1(1g),乙腈(50ml),boc胺(1.5g),tfa(2.2g),三乙基硅烷(7.4g),室温搅拌反应过夜;取样检测,大部分为原料;补加0.5g boc胺,继续搅拌反应;取样检测,基本无变化;将反应液减压浓缩干,pe:ea=10:1柱层析,tlc点挨得较近,得不纯产物2g。直接进行下一步反应。
[0024]
化合物3的合成:
[0025]
向100ml单口瓶中加入化合物2(2g),dcm(20ml),m-cpba(4g),室温搅拌反应过夜;取样检测,原料基本无剩余;向反应液加入20ml碳酸氢钠水溶液,搅拌10min;分液,水相用dcm(20ml)萃取一次;合并有机相,无水硫酸钠干燥;滤去干燥剂,滤液减压浓缩干,pe:ea=8:1爬大板,得0.7g黄色油状物。直接进行下一步反应。
[0026]
化合物4的合成:
[0027]
向100ml单口瓶中加入化合物3(0.7g),二氧六环(10ml),sm3(1.2g),k2co3,(1.88g,3eq),pd(pph3)4(0.05g,0.01eq),氮气置换气体三次;升温至90-95℃搅拌反应;取样检测,原料基本无剩余;将反应液减压浓缩干,柱层析提纯。通过相同方法将化合物的总量合成到5g。
[0028]
化合物5的合成:
[0029]
向100ml三口瓶中加入化合物4(6.3g),再加入hcl/meoh,搅拌反应;tlc检测原料基本转化完全;减压将反应液浓缩至干,直接用于下一步;
[0030]
化合物iii的合成:
[0031]
50ml三口中加入化合物5(4.2g),sm4(4.18g),diea(4.07g),meoh(80ml),搅拌升温至回流,搅拌过夜;将反应液降温至室温,过滤出固体,仅有量产物,滤液检测,将滤液减压浓缩至干。ms(esi)m/z=435.5[m h]
。
[0032]
实施例4化合物ii的制备
[0033][0034]
单口瓶中将rex-p25-9(1g,2.38mmol)溶于二氯甲烷(10ml)中,将反应液用冰水浴降温至0℃,缓慢加入m-cpba(3eq,1.23g,7.15mmol)。体系在室温中反应2h,爬小板(dcm:meoh=15:1)显示rex-p25-9原料点消失。将反应液倒入30ml的冰水中,用二氯甲烷(10ml)萃取三次,饱和食盐水洗涤一次,无水硫酸镁干燥,过滤除去固体,旋转蒸发除去溶液,得到白色不溶物固体化合物ii(912mg)。ms(esi)m/z=451.5[m h]
。
[0035]
实施例5化合物iv的制备
[0036][0037]
化合物2的合成:
[0038]
将化合物1(1.07g,7.0mmol)溶解在1,2-二氯乙烷(10ml)中,分别加入nbs(1.62g,9.1mmol)和偶氮二异丁腈(92mg,0.56mmol)。将体系加热到85℃反应30mins。tlc显示反应完成。将反应液倒入30ml水中,用二氯甲烷(10ml)萃取三次,饱和食盐水洗涤一次,无水硫酸镁干燥,过滤除去固体,旋转蒸发除去溶液,使用层析柱提纯(pe/ea=80:1)得到白色固体目标产物2(750mg,3.24mmol,47%yield)。ms(esi)m/z 230.7[m h]
。
[0039]
化合物3的合成:
[0040]
向dmf(4ml)分别加入化合物2(350mg,1.5mmol),k2co3(420mg,3.04mmol),acoh(137mg,2.3mmol)缓慢滴加进反应液中。将体系在室温下反应2h,tlc显示反应完成。将反应液倒入30ml水中,用乙酸乙酯(10ml)萃取三次,饱和食盐水洗涤一次,无水硫酸镁干燥,过滤除去固体,旋转蒸发除去溶液,使用层析柱提纯(pe:ea=10:1)得到白色固体目标产物(300mg,95%yield)。ms(esi)m/z 210.9[m h]
。使用同样的方法合成化合物总量为800mg后进行下一步反应。
[0041]
化合物4的合成:
[0042]
将化合物3(800mg,3.79mmol),2-甲基-4-吡啶硼酸,k2co3(1.05g,5.78mmol)和pd(dppf)cl2ch2cl2(155mg,0.19mmol)混合溶解在二氧六环和水的混合溶剂22ml(二氧六环:
水=10:1)中。在氮气保护下于120℃反应过夜。tlc显示反应完成。将反应液倒入60ml水中,用乙酸乙酯(20ml)萃取三次,饱和食盐水洗涤一次,无水硫酸镁干燥,过滤除去固体,旋转蒸发除去溶液,使用层析柱提纯(dcm:meoh=100:1)得到淡黄色固体目标产物(210mg,0.933mmol,25%yield)。ms(esi)m/z 226.0[m h]
。
[0043]
化合物5的合成:
[0044]
将化合物4(210mg,0.930mmol)和雷尼镍(240mg)溶解在甲醇(12ml)和四氢呋喃(3ml)的混合溶剂中,在氢气环境下室温反应过夜。tlc显示反应完成。过滤,旋转蒸发除去溶液,得到目标产物粗品(205mg),粗品目标产物直接进行下一步反应。ms(esi)m/z 230.1[m h]
。
[0045]
化合物7的合成:
[0046]
将化合物6(252mg,1.00mmol)溶解在甲醇(7.5ml)中,在0℃的冰浴条件下缓慢逐滴加入氢氧化锂水溶液(44mg,in 2.5ml h2o)。体系在0℃的冰浴条件下反应1h。旋转蒸发除去溶液,得到目标产物粗品(236mg),粗品目标产物直接进行下一步反应。ms(esi)m/z 226.0[m h]
。
[0047]
化合物iv的合成
[0048]
将化合物7(236mg,crude),hatu(570mg,1.50mmol)和et3n(150mg,1.50mmol)混合在dmf(4ml)中,体系在室温下搅拌5mins。化合物5(205mg,crude)溶解在dmf(2ml)中逐滴加入到反应体系中。将体系在氮气保护下室温反应过夜。反应结束后使用mplc提纯得到最终产物化合物iv。(57.6mg,0.132mmol,15%yield)。ms(esi)m/z=436.1[m h]
。1h nmr(400mhz,dmso-d6)δ10.04(t,j=6.0hz,1h),8.6(s,1h),8.52(d,j=4.8hz,1h),8.01(s,1h),7.93(d,j=7.6hz,2h),7.65(dd,j=14.4,7.8hz,1h),7.54
–
7.45(m,1h),7.44(s,1h),7.38(d,j=4.4hz,1h),5.46(t,j=5.2hz,1h),4.61(d,j=5.6hz,2h),4.50(d,j=5.2hz,2h),2.53(s,3h)。
[0049]
实施例6 super-top-flash(stf)报告基因试验
[0050]
化合物采用稳定转染stf报告基因的hek293t-stf细胞株同l-wnt3a分泌细胞株共培养方式测定其对wnt信号通路的抑制活性,该活性采用ic
50
这一指标来表示,ic
50
即stf报告基因表达的luciferase活性被抑制50%时的化合物的浓度。
[0051]
本发明实施例制备的化合物,利用crownbio公司的wnt报告基因平台进行测定,测定结果见表一。结果表明,本发明提供的化合物从分子水平上有较好的wnt信号通路抑制活性。
[0052]
表一 实施例化合物对wnt通路stf报告基因抑制的活性测定
[0053]
化合物ic
50
(nm)化合物i3.563化合物v2.11
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

