化合物作为sirt1受体激动剂的应用
技术领域
1.本发明属于药品或保健品领域,具体涉及一种化合物作为sirt1受体激动剂的应用。
背景技术:
2.在化学医药领域中,尿酸是人类嘌呤化合物的最终代谢产物。嘌呤代谢紊乱导致高尿酸血症。在正常嘌呤饮食状态下,非同日两次空腹血尿酸水平男性高于420μmol/l,女性高于360μmol/l,即称为高尿酸血症(hyperuricemia)。通常,单纯的处于高尿酸血症状态并没有自觉症状,但如果长时间处于该状态,血液中的尿酸盐将发生结晶,沉积在关节、皮下组织、肾脏等部位,进而出现痛风及痛风并发症等一系列临床表现。在最近公布的《2017年中国痛风现状报告白皮书》中显示,我国高尿酸血症患者人数已达1.7亿,其中痛风患者超过8000万人,而且正以每年9.7%的年增长率迅速增加。预计2020年,痛风人数将达到1亿。现今痛风已经成为我国仅次于糖尿病的第二大代谢类疾病,并严重危害着人们的生命和健康。
3.目前,治疗高尿酸血症、高尿酸血症引起的痛风及痛风并发症时,需要对血液中的尿酸进行控制:对于尿酸排泄不良型患者(占90%)适用促进尿酸排泄的药物,如:苯溴马隆、雷西纳德等;对于尿酸生成过多型患者适用抑制尿酸生产的药物(主要为黄嘌呤氧化酶抑制剂),如:别嘌醇、非布司他。但随着这些药物临床应用的增加,其不良反应也逐渐暴露。
4.别嘌呤醇(allopurinol)是最早上市的抑制尿酸生成的药物,自1963年应用于临床以来,因其价格低廉、降尿酸效果好,一直是治疗慢性痛风的主要药物。但随着别嘌呤醇的推广,不良反应的报道也逐渐增多,从20世纪70年代开始就有报道表明别嘌呤醇可引起肝肾损伤、白细胞减低、皮疹等不良反应,有约1.5%的过敏风险,严重的可能发生致死性过敏,引起了全世界范围的关注。因此,为减少不良反应,对于别嘌呤醇需从小剂量起用。
5.非布司他(febuxostat,商品名:uloric,武田北美制药公司)是一种非嘌呤类选择性黄嘌呤氧化酶抑制剂,于2008年5月在欧盟上市,2009年3月经美国fda批准上市,2013年进入中国市场,用于长期治疗伴随痛风的高尿酸血症。相较于其他治疗高尿酸血症的药物,非布司他具有更高的选择性和更强的活性。但是,相关研究及临床实践表明,非布司他也具有一定的不良反应:常见的不良反应有肝功能异常(3.5%)、腹泻(2.7%)、头痛(1.8%)、恶心(1.7%)和皮疹(1.5%)等。2017年11月15日,fda发布了非布司他心脏相关性死亡风险警告;2018年2月7日,cfda发布药物警戒快讯“一项由6000例痛风患者参与的安全性临床试验的初步结果表明,与别嘌醇相比,非布司他可能增加心脏相关性死亡的风险”。
6.上述药物常规剂量下毒副作用较大,加之上述药物耐受性普遍较低,在一定程度上限制了这些药物的临床应用。
7.沉默信息调节因子1(silent information regulator 1,sirt1)为sirtuin蛋白家族成员之一,存在于真核和原核生物中。其在细胞存活、增殖过程中发挥着重要的生理作用,其功能失调涉及生理活性如衰老,以及多种疾病,如肿瘤、糖尿病、心血管疾病、慢性炎
c7的烯基、rm取代或未取代的c
1-c7的酯基;
19.rm选自羟基、醛基、羧基、酯基,以及由中的至少一种所形成的糖基残基。
20.进一步地,r1为oh,r2和r3独立地选自h、oh、独立地选自h、oh、独立地选自h、oh、且r2和r3不同时为h。
21.进一步地,r1为h,r2和r3独立地选自h、oh、ch3、、r2和r3不同时为h。
22.进一步地,r1为或者r2和r3独立地选自h、oh、ch3、
r2和r3不同时为h。
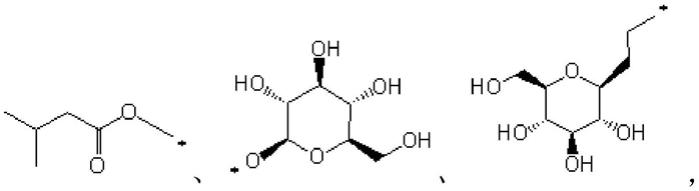
23.进一步地,式(i)所示的化合物选自:
24.25.[0026][0027]
进一步地,所述的应用包括制备预防和/或治疗与sirt1受体功能失调相关的疾病的药物。
[0028]
进一步地,与sirt1受体功能失调相关的疾病优选为衰老、肿瘤、糖尿病、心血管疾病、慢性炎症、肥胖、高尿酸血症、痛风。
[0029]
进一步地,所述慢性炎症为类风湿关节炎、慢性肾衰、痛风性关节炎。
[0030]
进一步地,其含有有效量的上述任一所述的一种或多种化合物、或其对映异构体、非对映异构体、盐、酯、前药、溶剂合物或盐的溶剂合物以及一种或多种药学上可接受的赋形剂。
[0031]
进一步地,所述药物为上述任一所述的一种或多种化合物、或其对映异构体、非对映异构体、盐、酯、前药、溶剂合物或盐的溶剂合物,按照常规工艺,加入常规辅料,制成临床上可接受的片剂、胶囊剂、散剂、合剂、丸剂、颗粒剂、糖浆剂、贴膏剂、栓剂、气雾剂、软膏剂或注射剂。
[0032]
进一步地,其含有有效量的上述任一所述的一种或多种化合物、或其对映异构体、非对映异构体、盐、酯、前药、溶剂合物或盐的溶剂合物以及一种或多种其他活性成分的组
合。
[0033]
本发明的技术方案具有如下优点:
[0034]
本发明通过sirt1活性测试发现,本发明制备的化合物均具有sirt1激活作用,可作为潜在的sirt1激动剂,通过高尿酸血症动物模型发现,本发明的化合物在体内具有显著的降尿酸作用,可以作为潜在的降尿酸或治疗痛风的药物。
具体实施方式
[0035]
本发明以下实施例和实验例中,化合物可以按照本发明实施例的方法进行制备获得,也可以按照现有技术文献中的方法制备获得。
[0036]
实施例1化合物1-1~1-8的制备
[0037][0038]
取菊科植物苍术5kg,粉碎,用40l甲醇加热回流提取,提取时间为两小时,提取两次,合并两次提取液,减压浓缩,得浓缩浸膏。将所得浓缩浸膏分散于3l水溶液中,然后用两倍体积的乙酸乙酯萃取两次,将所得乙酸乙酯层减压浓缩,得到508g提取物,将该提取物分散于正己烷中,超声溶解20分钟,随后离心,得到沉淀120g。将该沉淀物溶解于500ml苯中,作为上样液,用硅胶柱进行分离纯化,以丙酮/苯溶液为洗脱液,按照丙酮与苯体积比分别为0:100、15:85、20:80、25:75、35:65、40:60进行梯度洗脱),上述各流动相均洗脱3bv,根据tlc检测结果进行合并洗脱液,分别保留丙酮体积百分数为15%、25%和35%的丙酮/苯洗
脱液进行后续纯化,其中将丙酮体积百分数为15%的丙酮/苯洗脱液使用反相硅胶柱进行纯化,用体积百分数45%甲醇水溶液作为洗脱液进行洗脱得到化合物1-1和1-2,将丙酮体积百分数为25%的丙酮/苯洗脱液使用反相硅胶柱进行纯化,用体积百分数38%甲醇水溶液作为洗脱液进行洗脱得到化合物1-4和1-5,将丙酮体积百分数为35%的丙酮/苯洗脱液使用反相硅胶柱进行纯化,用体积百分数30%甲醇水溶液作为洗脱液进行洗脱得到化合物1-6和1-7。
[0039]
取100mg化合物1-2,溶于350ml的3%氢氧化钾甲醇溶液中,60℃加热10分钟,待反应液冷却至室温后,加入1n的hcl溶液进行中和,减压浓缩除去反应液中的甲醇得到浓缩混合液,浓缩混合液再使用2倍体积乙醚萃取两次,所得有机层萃取物加入无水na2so4,即得到化合物1-3。取100mg化合物1-6,溶于350ml的3%氢氧化钾甲醇溶液中,60℃加热10分钟,待反应液冷却至室温后,加入1n的hcl溶液进行中和,减压浓缩除去反应液中的甲醇得到浓缩混合液,浓缩混合液再使用2倍体积乙醚萃取两次,所得有机层萃取物加入无水na2so4,即得到化合物1-8。
[0040]
将上述制备得到的化合物分别通过1h-nmr、
13
c-nmr和hplc-ms进行结构确认,结构确认数据参考如下文献:
[0041]
化合物1-1~1-3:nakai y,sakakibara i,hirakura k,et al.a new acetylenic compound from the rhizomes of atractylodes chinensis and its absolute configuration.[j].cheminform,2006,37(19):1580-1581.
[0042]
化合物1-4~1-5:xu k,feng z m,jiang j s,et al.sesquiterpenoid and c14-polyacetylene glycosides from the rhizomes of atractylodes lancea[j].chinese chemical letters,2017,28(3):597-601.
[0043]
化合物1-6~1-8:yoshihiro kano,ken-ichi komatsu,et al.a new polyacetylene compound from atractylodes rhizome[j].chem pharm bull,1989,37(1):193—194.
[0044]
实施例2化合物2-1~2-7的制备
[0045][0046]
取干燥的菊科鬼针草60kg,加入5倍重量的体积百分数为70%乙醇水溶液,室温浸泡提取三天,提取两次,合并提取液,减压浓缩去除乙醇,得浸膏。将所得浸膏用hpd-100大孔树脂进行柱层析,用纯水和体积百分数为70%的乙醇水溶液进行洗脱,将所得的70%乙醇洗脱液减压浓缩得到2.1kg固体。将该固体用d101大孔树脂进行柱层析,依次用纯水,20%,35%,60%,95%乙醇水溶液进行洗脱,上述各流动相均洗脱3bv,收集60%乙醇洗脱液,减压浓缩得到浸膏。将该浸膏使用硅胶柱层析进行分离,氯仿甲醇梯度洗脱(氯仿与甲醇的体积比100:1、50:1、35:1、20:1、10:1、5:1、3:1、2:1、1:1的混合溶液,上述各流动相均洗脱3bv)收集到8个组分fr6.1-fr6.8,其中fr6.2使用sephadex lh-20柱层析分离,使用甲醇水溶液梯度洗脱(10%,25%,40%,60%,100%甲醇水溶液),上述各流动相均洗脱3bv,得到fr6.2-1至fr6.2-5,其中fr6.2-2使用硅胶柱色谱进行纯化,以石油醚和乙酸乙酯的混合液为流动相进行洗脱(石油醚:乙酸乙酯=10:1)得到化合物2-5。组分fr6.2-3使用反相硅胶制备柱色谱进行分离,使用甲醇水溶液梯度洗脱(20%,30%,45%,60%,95%甲醇水溶液),上述各流动相均洗脱3bv,得到组分fr6.2-3-1至fr6.2-3-5,其中fr6.2-3-2使用硅胶柱层析纯化,以石油醚乙酸和乙酯溶液的混合液洗脱(石油醚:乙酸乙酯=2:1)得到化合物2-1,其中fr6.2-3-3使用硅胶柱层析纯化,以石油醚和乙酸乙酯溶液的混合液洗脱(石油醚和乙酸乙酯的体积比为5:1)得到化合物2-3。fr6.5使用sephadex lh-20柱层析分离,使用甲醇水溶液梯度洗脱(10%,15%,30%,45%,55%,70%,100%甲醇水溶液),上述各流
动相均洗脱3bv,得到fr6.5-1至fr6.5-7,其中fr6.5-3使用反相硅胶制备柱色谱进行分离,使用甲醇水溶液梯度洗脱(30%,40%,50%,60%,95%甲醇水溶液),上述各流动相均洗脱3bv,其中40%甲醇水溶液洗脱组分减压浓缩得到化合物2-2,50%甲醇水溶液洗脱组分减压浓缩得到化合物2-4。fr6.7使用反相硅胶制备柱色谱进行分离,使用甲醇水溶液梯度洗脱(35%,45%,55%,65%,95%甲醇水溶液),上述各流动相均洗脱3bv,45%甲醇水溶液洗脱组分减压浓缩得到化合物2-6,65%甲醇水溶液洗脱组分减压浓缩得到化合物2-7。
[0047]
将上述制备得到的化合物分别通过1h-nmr、
13
c-nmr和hplc-ms进行结构确认,结构确认数据参考如下文献:
[0048]
化合物2-1~2-6:wang x y,chen g r,pan c x,et al.polyacetylenes from bidens bipinnata l.and their biological activities[j].phytochemistry letters,2014,7:198-201.
[0049]
化合物2-7:jiamei l,wenquan l,xiaojuan x,et al.anti-inflammatory constituents from bidens frondosa[j].molecules,2015,20(10):18496-18510.
[0050]
实施例3化合物3-1~3-5的制备
[0051][0052]
取干燥长毛风毛菊全草20kg,用5倍倍重量的体积浓度为70%乙醇水溶液室温浸泡提取,每次浸泡提取三天,提取两次,合并两次提取液,减压浓缩去除乙醇,得到浸膏,所得浸膏用两倍体积乙酸乙酯萃取两次,合并两次乙酸乙酯层萃取液,减压浓缩得到粗提取物,将该粗提取物经过硅胶拌样,使用硅胶柱层析,分别以正己烷:乙酸乙酯(体积比分别为25:1、20:1、15:1、10:1、7:1、4:1、2:1、1:1),以及乙酸乙酯作为流动相梯度洗脱,上述各流动相均洗脱3bv,根据薄层色谱(tlc)检测结果进行合并,得到5个组分fra~fre。将frb使用sephadex lh-20柱层析分离,使用甲醇水溶液梯度洗脱(10%,20%,30%,40%,50%,70%,85%,100%甲醇水溶液),上述各流动相均洗脱3bv,根据薄层色谱(tlc)检测结果进行合并,得到5个组分frb-1~frb-5,其中frb-2使用agilent sd-1反相制备液相色谱进行分离,以甲醇水溶液为流动相进行梯度洗脱(15%,25%,30%,40%,50%,75%,95%甲醇水溶液),上述各流动相均洗脱3bv,得到化合物3-1,3-2。frb-3重复frb-2的分离步骤,得到化合物3-3。frb-4重复frb-2的分离步骤得到化合物3-4。frc使用sephadex lh-20柱层析分离,使用甲醇水溶液梯度洗脱(15%,30%,40%,50%,60%,70%,85%,100%甲醇水溶
液),上述各流动相均洗脱3bv,根据薄层色谱(tlc)检测结果进行合并,得到4个组分frc-1~frc-4,frc-1使用agilent sd-1反相制备液相色谱进行分离,以30%甲醇水溶液为流动相进行等度洗脱,得到化合物3-5。
[0053]
将上述制备得到的化合物分别通过1h-nmr、
13
c-nmr和hplc-ms,紫外全波长扫描及红外图谱进行结构确认,结构确认数据参考如下文献:
[0054]
化合物3-1:刘超,窦德强.于潜白术的化学成分研究[j].中华中医药学刊,2014(7):1615-1617.
[0055]
化合物3-2:zhang n,liu c,sun t m,et al.two new compounds from atractylodes macrocephala with neuroprotective activity[j].jourmal of asian natural products research,2017,19(1):35-41.
[0056]
化合物3-3:anet e f l j,lythgoe b,silk m h,et al.62.oenanthotoxin and cicutoxin.isolation and structures[j].jourmal of the chemical society(resumed),1953:309.
[0057]
化合物3-4~3-5:christensen l p,j rgen lam.flavones and other constituents from centaurea species[j].phytochemistry,1991,30(8):2663-2665.
[0058]
实施例4化合物4-1~4-13的合成
[0059][0060]
步骤a):取5mmo原料7,10mmolcbr4和20mmolpph3,150mlch2cl2混合,室温反应4小时,得到化合物8;
[0061]
步骤b):取5mmo原料9,5mmolbuli和50mlthf混合,置于-78℃,5分钟,0℃,10分钟,-78℃,加入5mmol i2室温反应10分钟,得到me3si-c≡c-i(化合物10);无需后续处理,立即使用;
[0062]
步骤c):向步骤a)的产物中加入11mmolmeli,50mlthf,-78℃,20分钟,室温10分钟,加入5mmol i2,从-78℃升温至室温,在室温下反应1小时,得到化合物11;
[0063]
步骤d):取5.5mmolme3si-c≡c-i,pd(dba)2(2mol-%),asph3(8mol-%),5mmo原料15和50mlthf混合,室温反应3小时,得到化合物12;
[0064]
步骤e):向步骤d)的产物中加入5mmol k2co3和10mlmeoh,室温反应14小时,得到化合物13;
[0065]
步骤f):取步骤c)的产物与步骤e)的产物混合,加入pd(dba)2(5mol-%),cui
(15mol-%)和80mlthf/ipr2nh(5:8,体积比),室温反应15分钟,得到化合物4-1(重复制备步骤a-f以得到足量的化合物4-1用于后续的实验);
[0066]
步骤g):向5mmol化合物4-1中加入5.5mmol dess-martin试剂和150mlch2cl2,室温反应5分钟,得到化合物4-2。
[0067]
将原料7分别更换为原料21或原料22或原料23进行步骤a)的反应,其他步骤不变,得到化合物4-3,4-4,4-5。
[0068]
将原料15更换为原料31进行步骤d)的反应,并且将原料7更换为原料21或原料22或原料23或原料24,其他步骤不变,得到化合物4-6,4-7,4-8,4-9。
[0069]
取1mmol化合物4-1,4-3,4-4,4-5分别溶于30ml无水ch2cl2中,加入全乙酰化溴代葡萄糖1.1mmol,于室温下搅拌30min,加入1.1mmol ag2co3,n2保护下,避光反应约24小时,过滤,滤液依次用30ml饱和nahco3,30ml饱和nacl,和30ml蒸馏水洗涤,无水mgso4干燥,减压浓缩,残余物溶于25ml无水甲醇中,加入甲醇钠0.5mmol,于室温反应30min,稀盐酸调节ph为6,减压浓缩,残余物经硅胶柱层析梯度洗脱,ch2cl2甲醇溶液梯度洗脱(7:1、12:1、20:1、30:1),上述各流动相均洗脱3bv,分别得到化合物4-10,4-11,4-12,4-13。
[0070]
[0086]1h-nmr(cdcl3):
[0087]
4.20(2h,d,j=5.2hz,5.6hz),5.91(1h,m),6.48(1h,dd,j=11.2hz,11.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.63(1h,d,j=15.6hz),6.69(1h,d,j=11.2hz),2.30(3h,s);
[0088]
13
c-nmr(cdcl3):122.6,79.9,77.5,77.5,79.9,110.9,137.6,197.7,140.9,129.5,127.3,65.6,28.3
[0089]
化合物4-6
[0090]
esi-ms 201[m]-[0091]1h-nmr(cdcl3):
[0092]
6.07(1h,d,j=15.6hz),7.47(1h,dd,j=11.2hz,11.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),5.71(1h,d,j=15.6hz),6.27(1h,m),4.20(2h,d,j=5.2hz,5.6hz);
[0093]
13
c-nmr(cdcl3):110.1,79.9,77.5,77.5,79.9,110.9,140.2,64.3,140.9,147.2,117.5,170.6
[0094]
化合物4-7
[0095]
esi-ms 215[m]-[0096]1h-nmr(cdcl3):
[0097]
6.07(1h,d,j=15.6hz),7.47(1h,dd,j=11.2hz,11.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.70(1h,d,j=15.6hz),6.43(1h,d,j=15.6hz);
[0098]
13
c-nmr(cdcl3):127.7,79.9,77.5,77.5,79.9,110.9,120.3,170.6,140.9,147.2,117.5,170.6
[0099]
化合物4-8
[0100]
esi-ms 213[m]-[0101]1h-nmr(cdcl3):
[0102]
6.07(1h,d,j=15.6hz),7.47(1h,dd,j=11.2hz,11.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.63(1h,d,j=15.6hz),6.69(1h,d,j=15.6hz),2.30(3h,s);
[0103]
13
c-nmr(cdcl3):122.6,79.9,77.5,77.5,79.9,110.9,137.6,197.7,140.9,147.2,117.5,170.6,28.3
[0104]
化合物4-9
[0105]
esi-ms 243[m]-[0106]1h-nmr(cdcl3):
[0107]
6.07(1h,d,j=15.6hz),7.47(1h,dd,j=11.2hz,11.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),5.71(1h,d,j=15.6hz),6.27(1h,m),4.75(2h,d,j=5.2hz),2.01(3h,s);
[0108]
13
c-nmr(cdcl3):110.1,79.9,77.5,77.5,79.9,110.9,140.2,140.9,147.2,117.5,170.6,65.8,170.3,20.8
[0109]
化合物4-10
[0110]
esi-ms 333[m]-[0111]1h-nmr(cdcl3):
[0112]
1.71(3h,dd,j=1.6hz,6.8hz),6.08(1h,m),5.50(1h,d,j=15.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.48(1h,dd,j=11.2hz,11.6hz),5.91(1h,m),4.04(2h,d,j=5.2hz),5.03(1h,d,j=6.8hz),3.73(1h,s),3.49(1h,s),3.40(1h,s),3.75(1h,s),3.54(2h,d,j=5.6hz);
[0113]
13
c-nmr(cdcl3):125.4,128.9,140.9,110.9,79.9,77.5,77.5,79.9,110.9,141.6,68.8,18,77.7,71.5,73.4,73.9,104.6,62.3
[0114]
化合物4-11
[0115]
esi-ms 349[m]-[0116]1h-nmr(cdcl3):
[0117]
4.20(2h,d,j=5.2hz),6.27(1h,m),5.71(1h,d,j=15.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.48(1h,dd,j=11.2hz,11.6hz),5.91(1h,m),4.04(2h,d,j=5.2hz),5.03(1h,d,j=6.8hz),3.73(1h,s),3.49(1h,s),3.40(1h,s),3.75(1h,s),3.54(2h,d,j=5.6hz);
[0118]
13
c-nmr(cdcl3):125.4,128.9,140.9,110.9,79.9,77.5,77.5,79.9,110.1,140.2,68.8,64.3,77.7,71.5,73.4,73.9,104.6,62.3
[0119]
化合物4-12
[0120]
esi-ms 363[m]-[0121]1h-nmr(cdcl3):
[0122]
6.43(1h,d,j=15.6hz),6.70(1h,d,j=15.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.48(1h,dd,j=11.2hz,11.6hz),5.91(1h,m),4.04(2h,d,j=5.2hz),5.03(1h,d,j=6.8hz),3.73(1h,s),3.49(1h,s),3.40(1h,s),3.75(1h,s),3.54(2h,d,j=5.6hz)
[0123]
13
c-nmr(cdcl3):125.4,128.9,140.9,110.9,79.9,77.5,77.5,79.9,127.7,120.3,68.8,170.6,77.7,71.5,73.4,73.9,104.6,62.3
[0124]
化合物4-13
[0125]
esi-ms 361[m]-[0126]1h-nmr(cdcl3):
[0127]
2.30(3h,s),6.69(1h,d,j=15.6hz),6.63(1h,d,j=15.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.48(1h,dd,j=11.2hz,11.6hz),5.91(1h,m),4.04(2h,d,j=5.2hz),5.03(1h,d,j=6.8hz),3.73(1h,s),3.49(1h,s),3.40(1h,s),3.75(1h,s),3.54(2h,d,j=5.6hz)
[0128]
13
c-nmr(cdcl3):125.4,128.9,140.9,110.9,79.9,77.5,77.5,79.9,122.6,137.6,68.8,197.7,77.7,71.5,73.4,73.9,104.6,62.3,28.3
[0129]
实施例5化合物5-1~5-3的制备
[0130][0131]
取菊科植物大丽花(dahlia pinnata cav)的干燥根茎5kg,粉碎后用3倍重量的体积浓度为70%的乙醇水溶液,室温下浸泡提取一周,提取两次,合并两次提取液,减压浓缩除去有机溶剂,得浸膏。将浸膏用1倍体积的乙醚萃取两次,将乙醚萃取液减压浓缩,得到胶状物,将该胶状物溶解于其10倍重量的汽油乙醚(体积比为9:1)溶液中,作为上样液,用硅胶柱分离,以汽油和乙醚的混合液为流动相,按照以下程序进行梯度洗脱,汽油与乙醚的体积比分别为9:1、7:1、5:1、3:1、1:1、1:3,最后用乙醚洗脱,上述各流动相均洗脱3bv,根据tlc检测结果合并洗脱液,得到混合物fr1-1。将混合物fr1-1再次用硅胶柱分离,以汽油和乙醚的混合溶液为洗脱液,按照如下程序进行梯度洗脱,汽油/乙醚的体积比为3:1、2:1、1:1、1:2、1:3、1:5,上述各流动相均洗脱3bv,根据tlc检测结果合并洗脱液,得到化合物1-9。
[0132]
步骤a):取1mmol化合物1-9,加入1.1mmoldess-martin试剂,30mlch2cl2,室温下反应5分钟,将所得产物经过反相硅胶制备柱色谱进行分离,以甲醇和水的混合液为洗脱液按照如下程序进行梯度洗脱,甲醇的体积百分数为20%、25%、30%、35%,上述各流动相均洗脱3bv,根据tlc检测结果合并洗脱液,浓缩干燥得到化合物1-10(重复该步骤以获取足够量的化合物1-10进行后续实验)。
[0133]
步骤b):取5mmol化合物1-10,6mmol三甲基氯化硅和60mln-甲基吗啉体系混合,室温反应8小时;
[0134]
步骤c):向步骤b)的混合液中加入5.5mmol的k2co3,10mlmeoh,室温,5.2mmoletmgbr,thf10ml,室温,随后加入5.5mmol的i2(溶于thf溶剂150ml),反应10分钟;
[0135]
步骤d):在-15℃下向步骤c)的产物中滴加6mmol的异丙基氯化镁-氯化锂的thf溶液(1.3m),搅拌2小时,将5mmol三甲基硅脂保护的葡萄糖酸内酯的正庚烷溶液缓慢滴加至体系中,升温至-10℃,反应3小时。滴加6mmol甲磺酸的甲醇溶液(0.62mol/l),自然升值室温,搅拌,反应8小时。滴加饱和碳酸氢钠溶液50ml淬灭反应,调整至ph7.5,石油醚萃取,旋蒸除去溶剂后得产物。
[0136]
步骤e):将前一步骤产物溶于乙腈和二氯甲烷的混合溶液60ml(体积比1:1),冷却至-8℃,加入三乙基硅烷10mmol,然后滴加三氟化硼乙醚络合物,7.5mmol,升温至0℃搅拌5小时,加入饱和碳酸氢钠50ml淬灭反应,调整ph7.5,减压浓缩除去有机溶剂,得到化合物5-1。将化合物1-10分别用化合物3-1和化合物3-4替代,其他步骤不变,可得化合物5-2,5-3。
[0137]
将上述制备得到的化合物分别通过1h-nmr、
13
c-nmr和hplc-ms进行结构确认:
[0138]
化合物1-9:结构确认数据参考如下文献::化合物1-1/1-2/1-3/1-5/1-6/1-7(bedford c t,bhattacharjee d,fairbrother j r f,et al.natural acetylenes.part xlix.polyacetylenes from dahlia scapigera(a.dietr.)link and otto var.scapigera f.scapigera and some dahlia hybrids.[j].chemischer informationsdienst,1976,7(25):735-741;
[0139]
化合物1-10:
[0140]
esi-ms:213[m] ;
[0141]
1h-nmr(cdcl3):6.87(1h,dd,j=15.6hz,10.8hz,c6-h),6.56(1h,d,j=15.6hz,10.8hz,c5-h),6.51(1h,d,j=15.6hz,c4-h),6.29(1h,d,j=6.4hz,c2-h),6.18(1h,dd,j=15.6hz,6.8hz,c13-h),5.78(1h,d,j=15.6hz,c7-h),5.56(1h,d,j=15.6hz,c12-h),1.77(3h,dd,j=6.8hz,1.6hz,c14-h);
[0142]
13c-nmr(cdcl3):c-1(196.4),c-2(109.1),c-3(189.0),c-4(122.5),c-5(130.8),c-6(140.3),c-7(110.9),c-8(80.2),c-9(76.4),c-10(72.41),c-11(81.3),c-12(110.41),c-13(141.6),c-14(18.67);
[0143]
化合物5-1:
[0144]
esi-ms:359[m]
[0145]1h-nmr(cdcl3):
[0146]
1.71(3h,dd,j=1.6hz,6.8hz),6.08(1h,m),5.50(1h,d,j=15.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.51(1h,dd,j=11.2hz,11.6hz),6.50(1h,d,j=15.6hz),6.21(1h,d,j=4.2hz),9.68(1h,d,j=4.2hz),4.36(1h,d,j=5.2hz),
3.44(1h,m),3.49(1h,m),3.40(1h,m),3.76(1h,m),3.79(1h,m),3.54(1h,m)
[0147]
13
c-nmr(cdcl3):135.2,130.4,140.9 110.9,79.9,77.5,79.9,110.9,141.6,168.7,127.2 191.1,18,81.9,71.6,77.6,75.5,89.7,62.4
[0148]
化合物5-2
[0149]
esi-ms 669[m]-[0150]1h-nmr(cdcl3):
[0151]
6.08(1h,m),5.50(1h,d,j=15.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.27(1h,dd,j=11.2hz,11.6hz),5.72(1h,dd,j=6.8hz,10.8hz),2.48(1h,m),3.70(1h,m),1.29(2h,dd,j=3.2hz,4.8hz),1.42(2h,dd,j=3.2hz,4.8hz),3.67(1h,m),3.71(1h,m),3.49(3h,m),3.40(6h,m),3.76(3h,m),3.79(3h,m),3.54(3h,m),2,21(2h,dd,j=4.8hz,6.4hz)
[0152]
13
c-nmr(cdcl3):137.4,128.8,140.9,110.9,79.9,77.5,77.5,79.9,109.4,146.3,41.2,24.4,81.8,71.5,77.5,73.4,71.4,62.3,28,81.4,71.5,77.1,75.5,82.7,62.3,32.8,81.5,71.5,77.2,75.6,78.3,62.3,
[0153]
化合物5-3
[0154]
esi-ms 403[m]-[0155]1h-nmr(cdcl3):
[0156]
1.71(3h,dd,j=1.6hz,6.8hz),6.08(1h,m),5.50(1h,d,j=15.6hz),6.87(1h,dd,j=11.2hz,11.6hz),5.74(1h,d,j=15.6hz),6.48(1h,dd,j=11.2hz,11.6hz),5.91(1h,d,j=15.6hz),4.64(1h,m),1.57(2h,m),1.42(2h,m),3.67(1h,m),3.44(1h,m),3.49(1h,m),3.40(1h,m),3.76(1h,m),3.79(1h,m),3.54(1h,m),2.01(3h,s)
[0157]
13
c-nmr(cdcl3):135.4,128.8,140.9,110.9,79.9,77.5,77.5,79.9,110.9,141.6,76.5,28.8,23.9,18,81.4,71.5,77.1,75.5,82.4,62.3,170.3,21.1。
[0158]
实施例6:
[0159]
前述实施例中制备的各化合物,按常规法加注射用溶媒,精滤,灌封灭菌后可制成注射液。
[0160]
实施例7:
[0161]
前述实施例中制备的各化合物,将其溶于无菌注射用水中,用无菌漏斗过滤,分装,低温冷冻干燥后无菌熔封即得粉针剂。
[0162]
实施例8:
[0163]
前述实施例中制备的各化合物,作为药物活性成分,使用几种常规赋形剂作为制备组合药物片剂或胶囊剂的辅料成分,按照常规方法制成每片或每粒胶囊含有药物成分0.5-300mg的片剂或胶囊剂。
[0164]
实验例1 sirt1活性测试
[0165]
本实验利用sirt1 fluorometric drug discovery kit(sirt1活性荧光定量检测试剂盒)并按照该试剂盒说明书的方法以检测化合物对sirt1体外活性的影响。配制含0.5u sirt1(在37℃1u=1pmol
·
min-1
)(除空白组外),1000μmol
·
l-1
nad
,100μmol
·
l-1
去乙酰酶底物,sirt1缓冲液(50mmol
·
l-1
tris-hcl,ph 8.0,137mmol
·
l-1
nacl,2.7mmol
·
l-1
kcl,1mmol
·
l-1
mgcl2,1mg
·
ml-1
bsa)的试验体系。实验组分别加入前述实施例制备的各个待检
测化合物,空白组与激动剂(白藜芦醇)或抑制剂(烟酰胺)对照组加入等量溶解化合物的溶剂;再加入已配好的试验体系,使加入待检测化合物与试验体系的总体积为25μl(待测化合物、激动剂和抑制剂的终浓度为200μm)。37℃条件下孵育30min后于每个试验孔中加入1
×
fluor de lys developer solution(含有2mmol
·
l-1
烟酰胺)25μl终止反应(烟酰胺终浓度为200μm),反应在96孔板中进行。多功能酶标仪360nm激发光,460nm发射光条件下测取各孔荧光值。
[0166]
化合物激活sirt1活性试验结果(n=3),应用单因素方差分析的统计学方法,计量数据以均数
±
标准差表示,与空白组比较,
*
表示和空白对照组相比p<0.05,
**
表示和空白对照组相比p<0.01,
***
表示和空白对照组相比p<0.001。
[0167]
表1化合物1-1~化合物3-5的sirt1相对活性(%,和空白组相比)
[0168]
组别相对活性%组别相对活性空白组100.0
±
4.2化合物2-1117.7
±
4.9
*
烟酰胺对照组62.1
±
4.7
***
化合物2-3121.3
±
6.4
**
白藜芦醇对照组125.2
±
5.2
**
化合物2-4118.6
±
6.6
**
化合物1-1117.9
±
5.5
*
化合物2-5139.5
±
7.2
***
化合物1-2128.5
±
4.9
**
化合物2-6123.9
±
6.9
**
化合物1-3123.6
±
6.3
**
化合物2-7122.8
±
5.1
**
化合物1-4122.6
±
6.0
**
化合物3-1135.5
±
7.2
***
化合物1-5120.7
±
5.4
**
化合物3-2122.2
±
4.9
**
化合物1-6147.7
±
6.1
***
化合物3-3118.8
±
6.9
**
化合物1-7142.6
±
4.3
***
化合物3-4137.1
±
7.8
***
化合物1-8139.1
±
5.2
***
化合物3-5144.4
±
7.2
***
[0169]
表2化合物4-1-化合物5-3的sirt1相对活性(%,和空白组相比)
[0170]
组别相对活性空白组100.0
±
4.2烟酰胺对照组62.1
±
4.7
***
白藜芦醇对照组125.2
±
5.2
**
化合物4-1146.3
±
4.8
***
化合物4-3124.7
±
7.3
**
化合物4-10120.8
±
6.9
**
化合物4-11121.7
±
5.6
**
化合物5-2139.4
±
6.2
***
化合物5-3128.8
±
7.0
**
[0171]
由上述结果可知,本发明制备的化合物均具有sirt1激活作用,可作为潜在的sirt1激动剂,且其中化合物1-6,1-7,1-8,2-5,3-1,3-4,3-5,4-1,5-2的相对活性和空白对照组相比p<0.001,对sirt1的激活作用更佳。
[0172]
实验例2本发明化合物降尿酸活性测试
[0173]
1、实验材料
[0174]
健康雄性km小鼠,体重为15-18g,由上海灵畅生物科技有限公司提供;按每笼5只
进行分笼处理后,在屏障系统内适应性饲养4天。
[0175]
2、实验方法
[0176]
2.1实验分组
[0177]
选取体重集中的小鼠按体重随机平均分组,每组10只,分别为空白对照组、模型对照组、阳性对照组、化合物组(具体化合物见结果汇总表格)。
[0178]
2.2给药方法
[0179]
适应期过后随即对小鼠进行灌胃给药,每天上午灌胃1次,连续灌胃给药7天。
[0180]
化合物组分别给予实施例制备的化合物30mg/kg,其中化合物分别用0.5%羧甲基纤维素钠(cmc-na)溶液进行混悬;阳性对照组给予非布司他1.0mg/kg,用相同体积的0.5%羧甲基纤维素钠(cmc-na)溶液进行混悬;空白对照组和模型对照组均用0.5%羧甲基纤维素钠(cmc-na)溶液灌胃;各组均连续灌胃给药7天。在第7天上午灌胃给药0.5小时后,对各组小鼠进行腹腔注射进行高尿酸血症造模。其中,空白对照组腹腔注射0.5%羧甲基纤维素钠(cmc-na)溶液;模型对照组、阳性对照组、化合物组均注射300mg/kg动物体重的氧嗪酸钾(oa),用cmc-na溶液进行溶解。
[0181]
3、实验数据检测与处理
[0182]
3.1检测指标
[0183]
高尿酸血症造模1.5小时后,各组小鼠摘除眼球进行采血,采血容量不低于0.5ml,血样采集后于室温放置约1小时,待血液完全凝固后于3500rpm/4℃条件下离心10分钟,取血清在同等条件下复离5分钟,然后取0.2ml血清通过生化分析仪检测ua值。
[0184]
3.2统计学分析
[0185]
用excel和spss对数据进行统计分析,计算平均数和sd,经单因素方差分析后比较各实验组的组间差异。
[0186]
4、实验结果
[0187]
给药7天后,各组对高尿酸血症小鼠的血清尿酸水平的影响如下表所示。
[0188]
表3对高尿酸血症小鼠血清中尿酸水平的影响(均值μmol/l)
[0189]
样品尿酸(μmol/l)空白对照组61.18模型对照组140.61
##
阳性对照组42.23
**
化合物1-649.97**化合物1-757.66**化合物1-855.27**化合物2-559.32
**
化合物3-188.98
*
化合物3-465.57
**
化合物3-582.33
*
化合物4-161.19
**
化合物5-262.44
**
[0190]
注:
##
表示和空白对照组相比,p<0.01;
**
表示和模型对照组相比,p<0.01;
*
表示和
模型对照组相比,p<0.05(t-test检验)
[0191]
由表3可知:
[0192]
(1)与空白对照组相比,模型对照组小鼠的血清中尿酸显著升高(p<0.01),这表明高尿酸血症模型造模成功;
[0193]
(2)与模型对照组相比,化合物组小鼠的血清中尿酸水平的降低具有显著性差异(p<0.01或p<0.05)。
[0194]
5、实验结论
[0195]
本发明化合物在体内具有显著的降尿酸作用,可以作为潜在的降尿酸的药物。
[0196]
根据文献[wang j,zhu x x,liu l,et al.sirt1 prevents hyperuricemia via the pgc-1α/pparγ-abcg2 pathway[j].endocrine,2016,53(2):443-452。]的报道,sirt1可促进回肠中abcg2的表达,从而促进肠道对于尿酸的排泄,并且还可以抑制肾脏中urat1的mrma水平,说明本发明化合物可能通过促进肠道和肾脏对尿酸的排泄来达到降低尿酸的效果。
[0197]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。