1.本发明涉及用于实时检测单一靶基因的遗传变异的单核酸及利用其的检测方法。更详细地,涉及利用单核酸实时检测单一靶基因的遗传变异的方法及用于该方法的试剂盒,上述单核酸具有x
‑
y
‑
z结构,并且由可与包含如单核苷酸多态性、点突变或mirna异构体(isoform)等的遗传变异的单一靶基因的一部分或全部碱基序列互补结合的碱基序列形成。
背景技术:
2.就转基因而言,因单核苷酸多态性(single nucleotide polymorphism;snp)而引起的变形为最常见的形态,并成为导致各种疾病的原因(barreiro lb,et al.,methods mol.biol.,578:255
‑
276,2009;beaudet l.et al.,genome res.,11(4):600
‑
608,2001)。对此,为了早期诊断出因转基因引起的各种疾病,通过snp检测的诊断方法非常有效且可实现快速诊断。因此,已经提出了许多用于准确检测snp的方法,并且至今还在进行着与之相关的许多研究(ermini ml.et al.,biosen.&bioele.,61:28
‑
37,2014;k.chang et al.,biosen.&bioele.,66:297
‑
307,2015)。
3.具体地,作为分析多个基因的最常用的方法,有使用聚合酶链式反应(polymerase chain reaction,pcr)的方法以及多重聚合酶链式反应(multiplex polymerase chain reaction,multiplex pcr)方法。
4.上述聚合酶链式反应可与模板(template)dna相结合,通过任意设计由荧光物质和猝灭物质结合而成的引物或探针,由此具有能够准确地仅扩增待检测的基因的所需位点的优点。但是,由于一次反应中只能扩增一个目的基因,因此具有待扩增的目的基因为多个时需要重复相同工作的麻烦。
5.上述多重聚合酶链式反应的优点在于,可在一个试管中进行多重聚合酶链式反应以同时分析多个基因区域。但是,随着在一个试管中同时使用多个引物或探针,在引物或引物之间发生交叉反应,因此一次可扩增的基因区域的数量受到限制。并且,寻找反应条件需要花费大量的精力和时间,并且具有无法从灵敏度及特异性获得较好的结果的缺点(hardenbol p.et al.,nat.biotechnol.,21(6):673
‑
678,2003)。
6.近来,正在积极开展着通过使用通用引物来同时扩增多个基因区域而不使用多重聚合酶链式反应以实现大量分析的研究,作为代表性技术,有可同时分析多个基因区域的单核苷酸多态性(snp)的snplex、goldengate assay、分子倒置探针(molecular inversion probes;mips)等。
7.上述snplex为如下的方法,即寡核苷酸连接分析(oligonucleotide ligation assay;ola)之后,进行利用核酸外切酶(exonuclease)的纯化过程,用探针(probe)两个末端的通用引物碱基序列进行聚合酶链式反应扩增之后,最后利用包含在探针(probe)的zipcode碱基序列来在dna芯片上进行分析(tobler et al.,j.biomol.tech.,16(4):398
‑
406,2005)。
8.goldengate assay为如下的方法,即利用上游(upstream)探针在固定于固体表面上的基因组dna(genomic dna)进行等位基因特异性引物延伸反应之后,与下游(downstream)探针进行dna连接反应,经过洗涤过程来除去未进行dna连接反应的多个探针之后,如snplex一样,用包含在探针中的通用引物碱基序列进行扩增,并在illumina珠芯片(illumina beadchip)上分析扩增的聚合酶链式反应产物(shen r.et al.,mutat.res.,573(1
‑
2):70
‑
82,2005)。
9.分子倒置探针(molecular inversion probes;mips)为如下的方法,即利用锁式探针(padlock probe)来进行缺口连接(gap
‑
ligation)之后,利用核酸外切酶(exonucelase)除去未进行dna连接的探针和基因组dna,利用尿嘧啶
‑
n
‑
糖基化酶(uracil
‑
n
‑
glycosylase)来使锁式探针线性化之后,利用包含在探针中的通用引物碱基序列来进行聚合酶链式反应,并将其与genflex tag array(affymetrix,艾菲矩阵公司)进行杂交以分析单核苷酸多态性(hardenbol p.et al.,nat.biotechnol.,21(6):673
‑
678,2003)。
10.然而,这些方法需要将在第一个试管中反应的生成物的一部分移至第二个试管中进行反应,或者需要利用各种酶,因此有可能发生不同样品之间的污染,具有实验方法复杂的问题。并且,由于仅可检测相当于附着有荧光标记的探针数量的单核苷酸多态性,因此存在随着待分析的单核苷酸多态性的数量增多而导致分析费用增加的问题。
11.点突变(point mutation)通常在dna复制中发生。当一个双链dna分子生成两个单链dna时发生dna复制,每条链用作生成互补链的模板。单点突变可改变整个dna序列,改变一个嘌呤(purine)或嘧啶(pyrimidine)可改变核苷酸编码的氨基酸。
12.点突变可发生在dna复制过程中的自发突变,并且由于这种原因,突变率会提高。
13.1959年,恩斯特
·
弗里兹(ernst freese)创造了术语“转换(transitions)”或“颠换(transversions)”来对不同类型的点突变进行区分。转换是用另一种嘌呤替代嘌呤碱或者用另一种嘧啶替代嘧啶。颠换是用嘧啶替代嘌呤,或者反之亦然。转换(alpha)及颠换(beta)在突变率有差异,通常转换突变大于颠换突变约10倍以上。
14.点突变的功能分类具有如无义突变,蛋白质的生成如stop
‑
gain及start
‑
loss一样异常缩短或增加的情况;如错义突变,编码其他氨基酸的情况(在braf基因中将缬氨酸(valine)改变为谷氨酸(glutamic acid)的情况,这将导致激活raf蛋白,从而在癌细胞中产生无限的增殖信号);如沉默突变,编码相同的氨基酸的情况,这些都是普通研究人员已知的事项。
15.众所周知,这种点突变为特定疾病的原因。多种肿瘤抑制蛋白中的点突变会导致癌症,而神经纤维瘤(neurofibromatosis)则是由neurofibromin1或neurofibromin 2基因的点突变引起的。镰状细胞性贫血(sickle
‑
cell anemia)是由血红蛋白的β
‑
珠蛋白链中的点突变引起的,其中在第6位,亲水性氨基酸谷氨酸被疏水性氨基酸缬氨酸替代。此外,它也被发现于泰伊
‑
萨克斯二氏病(tay
‑
sachs disease)或色盲中。
16.这种点突变的分析尤其在诊断癌症选择治疗剂中特别重要。就伴随诊断而言,根据癌症突变选择优化的抗癌药或排除特定抗癌药作为重要的判断依据,最近,靶向特定突变的抗癌药的开发一直在持续增加。
17.这种突变的分析方法通过pcr、ngs、ddpcr等实现,并且液体活检的重要性和关注
也持续增加,因此需要更加精确的测定方法。但是,目前已知的方法没有足够的分析能力,或者具有足够的分析能力时还要求具备昂贵的额外设备和复杂的分析过程,因此需要开发可更加简单地分析的方法。
18.对此,长期以来,在实时检测snp、点突变(point mutation)、mirna的异构体(isoform)等转基因的方面上,本发明人一直在研究可通过提高低灵敏度和特异性来简单而准确地诊断癌症的方法,本发明人发现,当使用单核酸时,它们具有高灵敏度及高特异性,并且对诊断包括癌症在内的因如癌症等的转基因而引起的各种疾病非常有效,并完成了本发明。
技术实现要素:
19.技术问题
20.本发明的一目的在于,提供单一靶基因的遗传变异检测用单核酸,其特征在于,上述单核酸:i)具有x
‑
y
‑
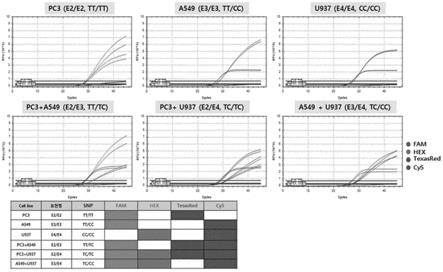
z结构,ii)与包含遗传变异的单一靶基因的一部分或全部碱基序列互补结合,iii)在两个末端或内部附着能够探测的相同或至少两个不同的标记,上述y为由位于单一靶基因的一个或两个碱基序列形成的rna。
21.本发明的再一目的在于,提供单一靶基因的遗传变异检测用单核酸,上述单核酸的特征在于,在单一靶基因的遗传变异为单核苷酸多态性(single nucleotide polymorphism;snp)的情况下,(a)上述x为由4个至20个碱基序列形成的dna,(b)上述z为由4个至20个碱基序列形成的dna。
22.本发明的另一目的在于,提供单一靶基因的遗传变异检测用单核酸,其特征在于,在单一靶基因的遗传变异为单核苷酸多态性(single nucleotide polymorphism;snp)或点突变(point mutation)或mirna异构体(isoform)的情况下,(c)上述x为由10至30个碱基序列形成的dna,(d)上述z为由1至5个碱基序列形成的dna。
23.本发明的还一目的在于,提供包含上述单核酸的单一靶基因的遗传变异实时检测用试剂盒。
24.本发明的又一目的在于,提供单一靶基因的遗传变异检测方法,上述方法包括:步骤a),从生物样品中获得包含待检测的遗传变异的靶核酸;步骤b),制备上述单一靶基因的遗传变异检测用单核酸;步骤c),混合从上述步骤a)中获得的靶核酸、在上述步骤b)中制备的单核酸、具有与上述步骤a)中获得的靶核酸互补的碱基序列的引物组及切断试剂之后,通过延伸反应来使包含遗传变异的靶核酸
‑
单核酸复合物扩增;以及步骤d),测定从上述步骤c)扩增的包含遗传变异的靶核酸
‑
单核酸复合物中分离的单核酸片段的量。
25.解决问题的方案
26.为了实现上述目的,本发明提供单一靶基因的遗传变异检测用单核酸。
27.详细地,上述单核酸:i)具有x
‑
y
‑
z结构,ii)与包含遗传变异的单一靶基因的一部分或全部碱基序列互补结合,iii)在两个末端或内部附着能够探测的相同或至少两个不同的标记,上述y为由位于单一靶基因的一个或两个碱基序列形成的rna,当单核酸与单一靶基因杂交时,y被切断试剂切断。
28.此时,在单一靶基因的遗传变异为单核苷酸多态性(single nucleotide polymorphism;snp)的情况下,上述单核酸的特征在于,(a)上述x为由4个至20个碱基序列
形成的dna,(b)上述z为由4个至20个碱基序列形成的dna,在单一靶基因的遗传变异为单核苷酸多态性(single nucleotide polymorphism;snp)点突变(point mutation)或mirna异构体(isoform)的情况下,上述单核酸的特征在于,(c)上述x为由10至30个碱基序列形成的dna,(d)上述z为由1至5个碱基序列形成的dna。
29.并且,在单一靶基因的遗传变异为单核苷酸多态性(single nucleotide polymorphism;snp)的情况下,上述单核酸的特征在于,单核酸与单一靶基因杂交之后,上述y被切断试剂切断时,上述x及z也从单一靶基因分离,并用作探针,在单一靶基因的遗传变异为单核苷酸多态性(single nucleotide polymorphism;snp)点突变(point mutation)或mirna异构体(isoform)的情况下,单核酸与单一靶基因杂交之后,上述y因切断试剂切断时,上述z从单一靶基因分离,但上述x不分离而同时用作引物及探针。
30.并且,本发明提供包含上述单核酸的单一靶基因的遗传变异实时检测用试剂盒。
31.并且,本发明提供单一靶基因的遗传变异检测方法,上述方法包括:步骤a),从生物样品中获得包含待检测的遗传变异的靶核酸;步骤b),制备上述单一靶基因的遗传变异检测用单核酸;步骤c),混合从上述步骤a)中获得的靶核酸、在上述步骤b)中制备的单核酸、具有与从上述步骤a)中获得的靶核酸互补性碱基序列的引物组及切断试剂之后,通过延伸反应来使包含遗传变异的靶核酸
‑
单核酸复合物扩增;以及步骤d),测定从上述步骤c)扩增的包含遗传变异的靶核酸
‑
单核酸复合物中分离的单核酸片段的量。
32.发明的效果
33.与现有的利用qpcr的snp及点突变(point mutation)分析方法相比,利用根据本发明的单核酸的实时检测单一靶基因的遗传变异的方法无需用于实时确认的额外的位置的探针(probe),因此具有可更加准确地测定如snp及点突变等的遗传变异的优点。即,根据本发明的单核酸仅备切断试剂切断,因此相比于利用现有探针的遗传变异检测方法可更加准确地进行测定。
34.并且,当利用根据本发明的单核酸来分析如snp及点突变之类的遗传变异时,可立即区分突变,而无需如解链温度(melting temperature)分析一样额外的突变确认过程。
35.因此,本发明的单核酸(promer)及利用其的实时检测单一靶基因的遗传变异的方法可快速而准确地区分在kras、egfr等中产生的各种点突变,从而可有效用于包括癌症在内的各种疾病的诊断、治疗剂选择以及预后诊断。
附图说明
36.图1为利用根据本发明一实施例的单核酸来确认apoe基因是否表达的图,详细地为用于确认可利用apoe单核酸1型(序列号3至6)来区分apoe基因6种的表型e2/e2、e3/e3、e4/e4、e2/e3、e2/e4、e3/e4的pcr结果。
37.图2为利用根据本发明一实施例的g13d单核酸1型来确认kras基因的g13d突变基因是否表达的图,详细地为将作为g13d突变细胞株的hct
‑
15细胞株的基因组dna(gdna)按照不同浓度稀释之后,利用g13d单核酸1型(序列号7)来确认根据gdna浓度的g13d突变基因是否表达的pcr结果。
38.图3为利用根据本发明一实施例的kras野生型单核酸来确认野生型kras基因的是否表达的图,详细地为将作为kras野生型细胞株的nci
‑
h1975细胞株的gdna按照不同浓度
稀释之后,利用kras野生型单核酸1型(序列号8)来确认根据gdna浓度的野生型kras基因是否表达的pcr结果。
39.图4为利用根据本发明一实施例的kras野生型单核酸1型来确认野生型kras基因是否表达的图。详细地为将作为hct
‑
15细胞株的基因组dna(gdna)按照不同浓度稀释之后,利用kras野生型单核酸1型(序列号8)来确认根据gdna浓度的野生型kras基因是否表达的pcr结果,该图为确认hct
‑
15细胞株是以杂合型(heterozygous type)的基因同时保留g13d突变和kras野生型的基因的细胞株的图。
40.图5为利用根据本发明一实施例的kras野生型单核酸1型来确认与野生型kras基因混合的kras基因g13d突变基因是否表达的图,详细地为将hct
‑
15细胞株的基因组dna(gdna)稀释为nci
‑
h1975细胞株的基因组dna(gdna)浓度的10%至0.01%,并与这些细胞株的基因组dna(gdna)混合之后利用g13d单核酸1型(序列号7)确认的pcr结果。
41.图6为利用根据本发明一实施例的单核酸2型来确认apoe基因是否表达的图,详细地为用于确认可利用apoe单核酸2型(序列号11至14)来区分apoe基因6种表型e2/e2、e3/e3、e4/e4、e2/e3、e2/e4、e3/e4的pcr结果。
42.图7为利用根据本发明一实施例的单核酸2型来确认kras基因的g12v突变基因是否表达的图,详细地为将作为g12v突变细胞株的sw620细胞株的基因组dna(gdna)按照不同浓度稀释之后,利用g12v单核酸2型(序列号15)来确认根据gdna浓度的g12v突变基因是否表达的pcr结果。
43.图8为利用根据本发明一实施例的单核酸2型来确认kras基因的g12c突变基因是否表达的图,详细地为将作为g12c突变细胞株的mia
‑
paca2细胞株的gdna按照不同浓度稀释之后,利用g12c单核酸2型(序列号16)来确认根据gdna浓度的g12c突变基因是否表达的pcr结果。
44.图9为利用根据本发明一实施例的单核酸2型来确认kras基因的g12s突变基因是否表达的图,详细地为将作为g12s突变细胞株的a549细胞株的gdna按照不同浓度稀释之后,利用g12s单核酸2型(序列号17)来确认根据gdna浓度的g12s突变基因是否表达的pcr结果。
45.图10为利用根据本发明一实施例的单核酸2型来确认egfr基因外显子20的t790m突变基因是否表达的图,详细地为将作为t790m突变细胞株的h1975细胞株和作为野生型细胞株的a549细胞株的gdna分别按照不同浓度稀释之后,利用t790m单核酸2型(序列号24)来确认根据gdna浓度的t790m突变基因是否表达的pcr结果。
46.图11为利用根据本发明一实施例的单核酸2型来确认let
‑
7a mirna基因是否表达的图,详细地为将通过rt
‑
pcr从let
‑
7a mirna合成而获得的let
‑
7a cdna按照不同浓度稀释之后,利用let
‑
7a单核酸2型(序列号21)来确认根据cdna浓度的let
‑
7a mirna基因是否表达的pcr结果。
47.图12为利用根据本发明一实施例的单核酸2型来确认let
‑
7d mirna基因是否表达的图,详细地为将通过rt
‑
pcr从let
‑
7d mirna合成而获得的let
‑
7d cdna按照不同浓度稀释之后,利用let
‑
7d单核酸2型(序列号24)来确认根据cdna浓度的let
‑
7d mirna基因是否表达的pcr结果。
48.图13为利用根据本发明一实施例的单核酸2型来确认let
‑
7mirna基因特异性检测
能力的图,详细地为在let
‑
7d cdna(1pm浓度)存在的情况下将let
‑
7a cdna按照不同浓度稀释之后,利用let
‑
7a单核酸2型(序列号21)来确认根据cdna浓度的let
‑
7a mirna基因是否表达,即,利用let
‑
7a单核酸2型(序列号21)来确认在let
‑
7d cdna存在的情况下的let
‑
7a mirna基因是否进行特异性检测的pcr结果。
49.图14为利用根据本发明一实施例的单核酸2型来确认mirna 34a基因是否表达的图,详细地为将通过rt
‑
pcr从mirna 34a合成而获得的mirna 34a cdna按照不同浓度稀释之后,利用mirna 34a单核酸2型(序列号27)来确认根据cdna浓度的mirna 34a基因是否表达的pcr结果。
50.图15为利用根据本发明一实施例的单核酸2型来确认mirna 34b基因是否表达的图,详细地为将通过rt
‑
pcr从mirna 34b合成而获得的mirna 34b cdna按照不同浓度稀释之后,利用mirna 34b单核酸2型(序列号30)来确认根据cdna浓度的mirna 34b基因是否表达的pcr结果。
51.图16为利用根据本发明一实施例的单核酸2型来确认mirna 34c基因是否表达的图,详细地为将通过rt
‑
pcr从mirna 34c合成而获得的mirna 34c cdna按照不同浓度稀释之后,利用mirna 34c单核酸2型(序列号33)来确认根据cdna浓度的mirna 34c基因是否表达的pcr结果。
52.图17为利用根据本发明一实施例的单核酸2型来确认mirna 34基因特异性检测能力的图,详细地为在mirna 34a cdna(100pm浓度)存在的情况下将mirna 34c cdna按照不同浓度稀释之后,利用mirna 34c单核酸2型(序列号33)来确认根据cdna浓度的mirna 34c基因是否表达,即,利用mirna 34c单核酸2型(序列号33)来确认在mirna 34a cdna存在的情况下的mirna 34c基因是否进行特异性检测的pcr结果。
53.图18为利用根据本发明一实施例的单核酸2型来确认mirna 34基因特异性检测能力的图,详细地为在mirna 34b cdna(100pm浓度)存在的情况下将mirna 34c cdna按照不同浓度稀释之后,利用mirna 34c单核酸2型(序列号33)来确认根据cdna浓度的mirna 34c基因是否表达,即,利用mirna 34c单核酸2型(序列号33)来确认在mirna34b cdna存在的情况下的mirna34c基因是否进行特异性检测的pcr结果。
54.图19为利用根据本发明一实施例的dna寡聚(dnaoligo)
‑
dna
‑
rna
‑
突变
‑
dna寡聚型的kras基因g12d的单核酸2型(g12d
‑
r1drmr2)来确认kras基因的g12d突变基因是否表达的图,详细地为将作为g12d突变细胞株的aspc
‑
1细胞株和作为kras野生型细胞株的ht
‑
29细胞株的gdna按照不同浓度稀释之后,利用g12d单核酸2型(序列号36)来确认根据gdna浓度的g12d突变基因表达的pcr结果。
55.图20为利用根据本发明一实施例的dna寡聚
‑
rna
‑
dna
‑
突变
‑
dna寡聚型的kras基因g12d的单核酸2型(g12d
‑
r1rdmr2)来确认kras基因的g12d突变基因是否表达的图,详细地为将作为g12d突变细胞株的aspc
‑
1细胞株和作为kras野生型细胞株的ht
‑
29细胞株的gdna按照不同浓度稀释之后,利用g12d单核酸2型(序列号37)来确认根据gdna浓度的g12d突变基因是否表达的pcr结果。
56.图21为利用根据本发明一实施例的dna寡聚
‑
dna
‑
突变
‑
rna
‑
dna
‑
dna寡聚型的kras基因g12d的单核酸2型(g12d
‑
r1dmrdr2)来确认kras基因的g12d突变基因是否表达的图,详细地为作为g12d突变细胞株的aspc
‑
1细胞株和作为kras野生型细胞株的ht
‑
29细胞
株的gdna分别按照不同浓度稀释之后,利用g12d单核酸2型(序列号38)来确认根据gdna浓度的g12d突变基因是否表达的pcr结果。
具体实施方式
57.本发明涉及单一靶基因的遗传变异实时检测用单核酸及利用其的检测方法。更详细地,涉及具有x
‑
y
‑
z结构,利用由可与包含如单核苷酸多态性(snp)、点突变或mirna异构体(isoform)等的遗传变异的单一靶基因的一部分或全部碱基序列互补结合的碱基序列形成的单核酸来实时检测单一靶基因的遗传变异的方法及用于该方法的试剂盒。
58.作为一实施方式,本发明提供单一靶基因的遗传变异检测用单核酸。
59.在本发明中,上述单核酸的特征在于,i)具有x
‑
y
‑
z结构,ii)与包含遗传变异的单一靶基因的一部分或全部碱基序列互补结合,iii)在两个末端或内部附着相同或至少两个不同的可探测标记。
60.此时附着的可探测的标记的位置不局限于特定位置,可使用通过切断试剂来切断y部位时可探测的标记分离的任何位置。
61.并且,上述单核酸的特征在于,与包含以实时检测为目的的遗传变异的单一靶基因的一部分或全部碱基序列互补结合来形成并扩增复合物。
62.在本发明中,上述单核酸是指用于探测遗传变异检测时的单核苷酸多态性(single nucleotide polymorphism;snp)、点突变(point mutation)或mirna异构体(isoform)的存在的核酸。其中,用于探测单核苷酸多态性(snp)的存在的单核酸可由“1型单核酸”或“单核酸1型”来表示,用于探测单核苷酸多态性(single nucleotide polymorphism;snp)或点突变或mirna异构体的存在的单核酸可由“2型单核酸”或“单核酸2型”来表示。不同于2型单核酸的上述1型单核酸可用作探针(probe),不用于1型单核酸的2型单核酸可同时用作引物(primer)和探针(probe)。
63.具体地,在1型单核酸与单一靶基因杂交之后,上述y通过切断试剂被切断的情况下,上述x及z也从单一靶基因分离并可用作探针,在2型单核酸与单一靶基因杂交之后,上述y通过切断试剂被切断的情况下,上述z虽然从单一靶基因分离,但上述x不分离,并同时用作引物和探针。
64.本发明的单核酸具有x
‑
y
‑
z结构,每个x、y及z可具有多种数量的核苷酸(nucleotide)。
65.上述y为位于单一靶基因的由一个或两个碱基序列形成的rna,并且为通过切断试剂被切断的位点。
66.其中,切断试剂优选地实现如dnase、rnase、解旋酶(helicase)、核酸外切酶及核酸内切酶等酶介导的切断,但也可使用其他已知的切断试剂。
67.在探测单核苷酸多态性(single nucleotide polymorphism;snp)的存在的情况下,上述x为由4个至20个碱基序列形成的dna,优选为由4个至19个碱基序列形成的dna,更优选为由4个至18个碱基序列形成的dna,进而优选为由5至18个碱基序列形成的dna,进而更优选为由6个至18个碱基序列形成的dna,进而更优选为由6个至17个碱基序列形成的dna,进而更优选为由6个至16个碱基序列形成的dna,最优选为由6个至15个碱基序列形成的dna。基于此的x属于1型单核酸。
68.并且,在探测单核苷酸多态性(single nucleotide polymorphism;snp)或点突变(point mutation)或mirna异构体(isoform)的存在的情况下,上述x为由10个至30个碱基序列形成的dna,优选为由11个至30个碱基序列形成的dna,更优选为由12个至30个碱基序列形成的dna,进而优选为由13个至30个碱基序列形成的dna,进而更优选为由14个至30个碱基序列形成的dna,进而更优选为由15个至30个碱基序列形成的dna,进而更优选为由15个至30个碱基序列形成的dna,进而更优选为由15个至29个碱基序列形成的dna,进而更优选为由15个至28个碱基序列形成的dna,进而更优选为由15个至27个碱基序列形成的dna,进而更优选为由15个至26个碱基序列形成的dna,进而更优选为由15个至25个碱基序列形成的dna,进而更优选为由15个至24个碱基序列形成的dna,进而更优选为由15个至23个碱基序列形成的dna,进而更优选为由15个至22个碱基序列形成的dna,进而更优选为由15个至21个碱基序列形成的dna,最优选为由15个至20个碱基序列形成的dna。基于此的x属于2型单核酸。
69.在探测单核苷酸多态性(single nucleotide polymorphism;snp)的存在的情况下,上述z为由4个至20个碱基序列形成的dna,优选为由4个至19个碱基序列形成的dna,更优选为由4个至18个碱基序列形成的dna,进而优选为由5个至18个碱基序列形成的dna,进而更优选为由6个至18个碱基序列形成的dna,进而更优选为由6个至17个碱基序列形成的dna,进而更优选为由6个至16个碱基序列形成的dna,最优选为由6个至15个碱基序列形成的dna。基于此的z属于1型单核酸。
70.并且,在探测点突变(point mutation)或mirna异构体(isoform)的存在的情况下,上述z为由1个至5个碱基序列形成的dna,优选为由2个至5个碱基序列形成的dna,更优选为由2个至4个碱基序列形成的dna,进而优选为由2个至3个碱基序列形成的dna。基于此的z属于2型单核酸。
71.在本发明中,上述遗传变异检测是指单核苷酸多态性(snp)、点突变(point mutation)或mirna异构体(isoform)的存在的探测,根据作为上述单核酸构成x及z的碱基数来特异而灵敏地探测它们每个遗传变异检测。
72.在本发明的一实施例中,通过实时pcr确认了可利用apoe单核酸(序列号3至6)来进行apoe基因6种表型e2/e2、e3/e3、e4/e4、e2/e3、e2/e4、e3/e4的明确区分(图1)。
73.在本发明的一实施例中,作为kras基因g12v突变细胞株的sw620细胞株的基因组dna(gdna)按照不同浓度稀释之后,利用g12v单核酸(序列号15)并通过实时pcr确认了根据gdna浓度的g12v突变基因是否表达(图7)。
74.在本发明的一实施例中,作为kras基因g12c突变细胞株的mia
‑
paca2细胞株的gdna按照不同浓度稀释之后,利用g12c单核酸(序列号16)并通过实时pcr确认了根据gdna浓度的g12c突变基因是否表达(图8)。
75.在本发明的一实施例中,作为kras基因g12s突变细胞株的a549细胞株的gdna按照不同浓度稀释之后,利用g12s单核酸(序列号17)并通过实时pcr确认了根据gdna浓度的g12s突变基因是否表达(图9)。
76.在本发明的一实施例中,将按照不同浓度分别稀释作为egfr基因外显子20的t790m突变细胞株的h1975细胞株和作为野生型细胞株的a549细胞株的gdna之后,利用t790m单核酸(序列号19)并通过实时pcr确认了根据gdna浓度的t790m突变基因是否表达
(图10)。
77.在本发明的一实施例中,let
‑
7a cdna按照不同浓度稀释之后,利用let
‑
7a单核酸(序列号21)并通过实时pcr确认了根据cdna浓度的let
‑
7a mirna基因是否表达(图11)。
78.在本发明的一实施例中,let
‑
7d cdna按照不同浓度稀释之后,利用let
‑
7d单核酸(序列号24)并通过实时pcr确认了根据cdna浓度的let
‑
7d mirna基因是否表达(图12)。
79.在本发明的一实施例中,在实验组中可以确认,向1pm浓度的let
‑
7d cdna2μl中,按各1/10添加将浓度从100fm稀释至1am的let
‑
7a cdna各2μl,可探测浓度为100fm至1fm的let
‑
7a cdna(图13)。
80.在本发明的一实施例中,按照不同浓度稀释mirna34a cdna之后,利用mirna 34a单核酸(序列号27)并通过实时pcr确认了根据cdna浓度的mirna 34a基因是否表达(图14)。
81.在本发明的一实施例中,按照不同浓度稀释mirna 34b cdna之后,利用mirna 34b单核酸(序列号30)并通过实时pcr确认了根据cdna浓度的mirna 34b基因是否表达(图15)。
82.在本发明的一实施例中,按照不同浓度稀释mirna 34c cdna之后,利用mirna 34c单核酸(序列号33)并通过实时pcr确认了根据cdna浓度的mirna 34c基因是否表达(图16)。
83.在本发明的一实施例中,在mirna 34a cdna或mirna 34b cdna中按照不同浓度稀释mirna 34c cdna之后,还可在微小浓度下,利用mirna 34c单核酸(序列号33)并通过实时pcr确认了mirna 34c基因是否表达(图17及图18)。
84.在本发明中,上述可探测标记可使用通过共价键或非共价键与单核酸结合的荧光物质或由荧光物质和猝灭物质形成的荧光对。
85.上述荧光物质可以为选自由例如cy3、cy5、cy5.5、bodipy、alexa 488、alexa 532、alexa 546、alexa 568、alexa 594、alexa 660、罗丹明(rhodamine)、tamra、fam、fitc、fluor x、rox、texas red、ornage green 488x、ornage green 514x、hex、tet、joe、oyster 556、oyster 645、bodipy 630/650、bodipy 650/665、calfluor ornage 546、calfluor red 610、quasar 670及生物素组成的组中的一种,但并不是必须局限于此。
86.并且,上述猝灭物质可以为选自由例如ddq
‑
1、dabcyl、eclipase、6
‑
tamra、bhq
‑
1、bhq
‑
2、bhq
‑
3、lowa black rq
‑
sp、qsy
‑
7、qsy
‑
2及mgbnfq组成的组中的一种,但并不是必须局限于此。
87.在本发明中使用荧光对作为可探测标记的情况下,荧光物质和猝灭物质的位置可以为位于x或z的形态或者位于y位点,但不限定于此。作为一例,荧光物质可位于x,猝灭物质可位于y或z。
88.本发明的单核酸作为单一靶基因的遗传变异,当检测点突变(point mutation)或mirna异构体(isoform)的存在时,可用作如下:i)用于从核酸中的rna合成cdna的rt引物;ii)用于扩增从核酸(dna或rna)合成的cdna的正向引物;iii)用于扩增从核酸(dna或rna)合成的cdna的反向引物;iv)用于扩增从核酸(dna或rna)合成的cdna正向引物及反向引物;或者v)用于实施确认待检测的核酸(dna或rna)的探针。
89.尤其,在将本发明的单核酸用作将rna(包含mirna等)合成及扩增为cdna的rt引物;或者正向引物和探针;或者反向引物和探针的情况下,合成cdna时,不需要将rt引物制造成环形或形成聚a的过程,可与待探测的rna杂交来合成cdna并扩增及实时检测待检测的rna(包含mirna等)。
90.作为另一实施方式,本发明提供包含上述单核酸的、用于实时检测单一靶基因的遗传变异的试剂盒。
91.当本发明的单核酸用作检测单一靶基因的遗传变异的试剂盒时,优选地,上述试剂盒除了本发明的单核酸以外还包含可切断单核酸的y位点的酶。
92.在本发明中,上述可切断单核酸的y位点的酶可任意使用可特异性切断单核酸的y位点的酶。例如,当y位点为dna时,优选地,使用dna核酸酶(dna nuclease,dnase),具体地使用dnaseⅰ、dnaseⅱ、s1核酸水解酶、核酸水解酶p1、ap核酸内部水解酶或uvrabsc核酸水解酶等,当y位点为rna时,优选地,使用rna水解酶(ribonuclease,rnase),具体地使用rnaseⅱ、rnaseⅲ、rnaseⅳ、rnaseh或rnase t2等。
93.当使用本发明的单核酸来用作检测单一靶基因的遗传变异的试剂盒时,上述试剂盒除了可切断本发明的单核酸及单核酸的y位点的酶以外,还可包含dna的扩增反应所需的试剂。
94.上述扩增反应所需的试剂可例举适量的dna聚合酶(例如,由thermusaquatiucs(taq)、thermusthermophilus(tth)、thermusfiliformis,thermis flavus、thermococcusliteralis或phyrococcusfuriosis(pfu)获得的热稳定性dna聚合酶)、dna聚合酶辅因子(mg
2
)、缓冲溶液、dntps(datp、dctp、dgtp及dttp)及水(dh2o)。并且,上述缓冲溶液有适量的曲拉通x
‑
100(triton x
‑
100)、二甲基亚砜(dimethylsufoxide;dmso)、tween20、nonidet p40、peg 6000、甲酰胺及牛血清白蛋白(bsa)等,但不限定于此。
95.作为又一实施方式,本发明提供单一靶基因的遗传变异检测方法,上述方法包括:步骤a),从生物样品中获得包含待检测的遗传变异的靶核酸;步骤b),制备上述的单核酸;步骤c),混合从上述步骤a)中获得的靶核酸、在上述步骤b)中制备的单核酸、具有与从上述步骤a)中获得的靶核酸互补的碱基序列的引物组及切断试剂之后,通过延伸反应来使包含遗传变异的靶核酸
‑
单核酸复合物扩增;以及步骤d),测定从上述步骤c)扩增的包含遗传变异的靶核酸
‑
单核酸复合物中分离的单核酸片段的量。
96.以下根据各个步骤来具体说明实时检测根据本发明的单一靶基因的遗传变异的方法。
97.步骤a),从生物样品中获得包含待检测的遗传变异的靶核酸。
98.在本发明中,包含上述遗传变异的靶核酸可以为从样品中待检测的rna或dna,或者可以为通过反转录聚合酶扩增上述rna而获得的cdna。
99.上述样品可以为生物样品或从生物样品中分离的rna、dna或它们的片段。具体地,上述样品可以为选自由血液、唾液、尿液、粪便、组织、细胞及活检标本组成的组中的一种以上,或者可以为从所保存的生物样品中分离的rna、dna或它们的片段。然而,并不是必须局限于此。
100.上述保存的生物样品为来自通过本领域通常已知的保存方法,保存一周以上、一年以上,例如,保存一年至十年,冷冻保存或者在常温下保存用福尔马林固定的组织的组织。
101.在本发明中,从样品中提取rna或dna的方法可利用本发明所属技术领域中所公知的多种方法。
102.步骤b),制备上述单核酸。
103.在本发明中,单核酸如上所述,详细而言,单核酸的特征在于,i)具有x
‑
y
‑
z结构,ii)与包含遗传变异的单一靶基因的一部分或全部碱基序列互补结合,iii)两个末端或内部附着相同或至少两个不同的可探测标记。
104.在本发明中,上述可探测标记可使用通过共价键或非共价键与单核酸结合的荧光物质或由荧光物质和猝灭物质形成的荧光对。
105.上述荧光物质可以为选自由例如cy3、cy5、cy5.5、bodipy、alexa 488、alexa 532、alexa 546、alexa 568、alexa 594、alexa 660、罗丹明(rhodamine)、tamra、fam、fitc、fluor x、rox、texas red、ornage green 488x、ornage green 514x、hex、tet、joe、oyster 556、oyster645、bodipy 630/650、bodipy 650/665、calfluor ornage 546、calfluor red610、quasar 670及生物素组成的组中的一种,但并不一定限于此。并且,上述猝灭物质可以为选自由例如ddq
‑
1、dabcyl、eclipase、6
‑
tamra、bhq
‑
1、bhq
‑
2、bhq
‑
3、lowa black rq
‑
sp、qsy
‑
7、qsy
‑
2及mgbnfq组成的组中的一种,但并不一定限于此。
106.步骤c),扩增包含遗传变异的靶核酸
‑
单核酸复合物。
107.在本发明中,包含上述遗传变异的靶核酸
‑
单核酸复合物的扩增可在混合上述获得的靶核酸、上述制备的单核酸、具有与上述获得的靶核酸互补的碱基序列的引物组及切断试剂之后,通过延伸反应来实现。
108.在本发明中,上述切断试剂优选地实现酶介导的切断,但也可使用其他公知的切断试剂。此时,使用术语“酶介导的切断(enzyme
‑
mediated cleavage)”以表示由如dnase、rnase、解旋酶(helicase)、核酸外切酶及核酸内切酶之类的酶催化的rna或dna的切断。在本发明的优选实施例中,优选地,杂交的探针的切口(nick)生成及切断由作为核酸内切酶或核酸外切酶的核糖核酸酶来执行。更优选地,核糖核酸酶为由双链dna
‑
rna杂交链在核糖核酸(ribonucleic acid)中生成切口而切断而成的双链核糖核酸酶。
109.在本发明中,上述切断试剂可以为rnaseh、rnaseⅱ、rnaseⅲ、rnaseⅳ或rnase t2的rna水解酶(ribonuclease,rnase),但不限定于此。
110.步骤d),测定从上述扩增的包含遗传变异的靶核酸
‑
单核酸复合物中分离的单核酸片段的量。
111.在本发明中,单核酸片段的量的测定可使用多种检测方法来进行。具体地,优选地,实时测定或反应结束之后测定根据本发明来分离的单核酸的片段,并且可通过荧光强度的变化或化学发光的测定来实现。
112.上述荧光强度的变化或化学发光的测定可使用本技术领域公知的可检测荧光标记的所有测定装置,例如,可使用triad多模式检测器(triad multimode detector)、wallac/victor荧光(wallac/victor fluorescence)或perkin
‑
elmer lb50荧光光谱仪(perkin
‑
elmer lb50bluminescence spectrometer)、lightcycler96、applied biosystems 7500或biorad cfx96 real
‑
time pcr thermocycler等来实现,但不限定于此。
113.根据本发明切断的单核酸片段的量的测定及检测方法可根据单核酸或流入反应液内的标记或可探测标记的种类而不同。
114.本发明的单核酸通过y位点切断之后扩增的步骤来使y位点的遗传变异区分变得容易,因此可通过此后的核酸扩增反应来确认突变。即,当本发明的单核酸中的y位点和靶
核酸的遗传变异位点实现杂交时,仅在y位点与遗传变异位点准确地互补结合的情况下才被切断,然后进行扩增反应,因此可明确确认是否发生遗传变异。具体地,当本发明的单核酸的y位点与靶核酸的遗传变异位点实现杂交时,若y位点未进行互补结合,则y位点不被切断,因此不引起扩增反应,这意味着靶核酸中不存在待测定的遗传变异。与此相反,若尽管在本发明的单核酸的y位点与靶核酸的发生遗传变异的位点实现非突变的情况和即使杂交,y位点也未互补结合的情况下,则y位点不被切断,因此不引起扩增反应,这意味着靶核酸中存在遗传变异。
115.在本发明中,上述遗传变异检测是指探测单核苷酸多态性(snp)、点突变(point mutation)或mirna异构体(isoform)的存在。
116.在本发明的一实施例中,按照不同浓度分别稀释作为g12d突变细胞株的aspc
‑
1细胞株和作为kras野生型细胞株的ht
‑
29细胞株的gdna之后,利用dna寡聚(dnaoligo)
‑
dna
‑
rna
‑
突变
‑
dna寡聚型的kras基因g12d的单核酸(g12d
‑
r1drmr2;序列号36),通过实时pcr确认了根据gdna浓度的g12d突变基因是否表达(图19)。
117.在本发明的一实施例中,按照不同浓度分别稀释作为g12d突变细胞株的aspc
‑
1细胞株和作为kras野生型细胞株的ht
‑
29细胞株的gdna之后,利用dna寡聚
‑
rna
‑
dna
‑
突变
‑
dna寡聚型的kras基因g12d的单核酸(g12d
‑
r1rdmr2;序列号37),通过实时pcr确认了根据gdna浓度的g12d突变基因是否表达(图20)。
118.在本发明的一实施例中,按照不同浓度分别稀释作为g12d突变细胞株的aspc
‑
1细胞株和作为kras野生型细胞株的ht
‑
29细胞株的gdna之后,利用dna寡聚
‑
dna
‑
突变
‑
rna
‑
dna
‑
dna寡聚型的kras基因g12d的单核酸(g12d
‑
r1dmrdr2;序列号38),通过实时pcr确认了根据gdna浓度的g12d突变基因是否表达(图21)。
119.在本发明中可容易使用的延伸反应,即核酸扩增反应是本发明所属技术领域的普通技术人员已知的。即,上述靶核酸的扩增包括聚合酶链式反应(polymerase chain reaction;pcr)、滚环扩增(rolling circle amplification;rca)、链置换扩增(strand displacement amplification;sda)或基于核酸序列的扩增(nucleic acid sequence based amplification;nasba),但上述方法不局限于此。上述核酸扩增产物为dna或rna。
120.通常,反应混合液中包含靶核酸、单核酸、核酸扩增反应的组成物及切口酶,使得可同时进行通过靶核酸的扩增及上述描述的单核酸的切断的探测。各个扩增反应需要分别单独地使缓冲液条件、引物、反应温度及单核酸切断条件等优化。结合核酸扩增反应来使用本发明的检测方法时,探测靶核酸的灵敏度和速度会显著提高。
121.另一方面,本发明的单一靶基因的遗传变异实时检测用单核酸可快速准确地检测及探测与癌症相关的kras、egfr等的点突变的基因,上述单核酸可适用于癌症诊断用试剂盒或癌症诊断用组合物,并且可有效利用于进行实时癌症诊断来提供是否产生癌症的信息。
122.以下,通过实施例来进一步详细说明本发明的结构及效果。这些实施例仅用于例示本发明,本发明的范围并不限定于这些实施例。
123.实施例1:利用1型单核酸的apoe分析
124.1型单核酸用于分析载脂蛋白e(apolipoprotein e,apoe)基因的6种表型e2/e2、e3/e3、e4/e4、e2/e3、e2/e4、e3/e4。
125.位于人类19号染色体的apoe基因为与心血管疾病及阿尔茨海默氏病有关的基因。通过密码子(codon)112(cys/arg)、密码子158(cys/arg)的dna的单核苷酸多态性(snp)[基因组dna位置第586(t/c)、第724(t/c)],apoe基因具有三种作为等位基因异构体(isoform)的apoeε2、apoeε3、apoeε4,并通过该等位基因的组合具有6种表型(e2/e2、e3/e3、e4/e4、e2/e3、e2/e4、e3/e4)。为了区分上述apoe基因的各个6种表型,可使用各个荧光染料(dye)所附着的4种改进形态的1型单核酸来通过4
‑
plex分析5'
‑
末端。现有分析方法中通常难以满足通过4
‑
plex的分析方法的灵敏度及特异性,但本发明中显示出可满足这些的结果。
[0126]
详细地,为了测定apoe基因的密码子112及密码子158的等位基因是否出现snp,如下表1所示,委托美国集成dna技术公司(integrated dna technologies;idt,usa)制备了根据本发明的1型单核酸和引物(primer)。其中,1型单核酸为具有x
‑
y
‑
z结构的探针,在5'
‑
末端分别附着了6
‑
fam、hex、texasred、cy5,并且在各个3'
‑
末端附着了iabkfq。并且,为了区别核糖核苷酸(rna)和脱氧核糖核苷酸(dna),在核糖核苷酸(rna)的序列前用角注“r”表示。
[0127]
表1
[0128]
apoe特异性引物组及4种apoe单核酸
[0129][0130]
为了分析,从韩国细胞株库分发了人细胞株pc3(e2/e2)、a549(e3/e3)、u937(e4/e4),从中获得了基因组dna(genomic dna)。为了分析6种表型,同型表型使用了pc3(e2/e2)、a549(e3/e3)、u937(e4/e4),异型表型以高浓度在每次反应中包含32ng(约104拷贝(copy))的方式将作为各个基因组dna的混合型的pc3 a549(e2/e3)、pc3 u937(e2/e4)、a549 u937(e3/e4)使用于分析。
[0131]
上述表1的apoe单核酸1、2、3、4的各0.2μm、0.15μm、0.15μm及0.075μm和apoe正向及反向引物的各0.35μm的最终浓度存在下,使用上述定量的基因组dna、0.5u rnase
‑
h、aptataq dna master(瑞士罗氏(roche)公司制造)4μl、gc rich solution(瑞士罗氏(roche)公司制造)3μl、无核酸酶的水(nuclease
‑
free water)将总体积(total volume)调节为20μl之后,进行聚合酶链式反应(polymerase chain reaction;pcr)。此时,pcr反应条件在95℃温度条件下5分钟,在95℃温度条件下15秒钟,在65℃温度条件下70秒钟,如此进行45个循环(cycle)。相关结果示于图1中。
[0132]
其结果,确认了可在一个反应孔(well)中分析6种apoe各个等位基因的组合。从该结果中可以确认,如先天突变(mutation)一样整个或只有一半发生突变(mutation)的情况下,使用改善的探针(probe)形态时区分能力优异。
[0133]
实施例2:利用1型单核酸的kras突变分析
[0134]
实施例2
‑
1:利用根据本发明的单核酸的实时kras基因的g13d突变分析
[0135]
为了测定利用根据本发明的1型单核酸来测定kras基因是否产生g13d突变(mutant),委托idt,制备了kras基因的g13d突变单核酸(1型)、kras野生型(wild type)单核酸(1型)、kras基因的g13d突变的正向及反向引物,其如下表2所示。此时,制备的单核酸在5'
‑
末端附着hex、fam,并在3'
‑
末端附着iabkfq。并且,为了区分核糖核苷酸(rna)和脱氧核糖核苷酸(dna),在核糖核苷酸(rna)的序列前用角注“r”表示。
[0136]
表2
[0137]
kras基因的g13d突变及野生型单核酸和g13d突变特异性引物组
[0138][0139][0140]
待测定的kras基因在从培养规定时间的hct
‑
15细胞株和nci
‑
h1975细胞株中提取的总基因组dna(total genomic dna)进行检测。总基因组dna使用purelink基因组dna迷你试剂盒(purelink genomic dna mini kit)(美国赛默飞世尔科技公司(thermo fisher scientific),cat no.k1820
‑
00)在各个细胞株的5
×
106细胞中提取,并利用nanodrop one(美国赛默飞世尔科技公司(thermo fisher scientific))进行定量。定量的总基因组dna稀释为15ng/μl,按各1/10进行系列稀释(serial dilution),使用15ng/μl~1.5pg/μl浓度的基因组dna各2μl。hct
‑
15细胞株为g13d突变细胞株,nci
‑
h1975已知为kras野生型细胞株。
[0141]
然后,准备上述制备的序列号7的10um浓度的g13d单核酸0.3μl,准备序列号9及10的10um浓度的多个引物各0.5μl。其中,向0.5u rnase
‑
h、aptataq dna master(瑞士罗氏(roche)公司制造)4μl、从nci
‑
h1975细胞株提取的总基因组dna30ng中分别添加从hct
‑
15细胞株提取的总基因组dna各30ng、3ng、300pg、30pg之后,用三次蒸馏水将总体积调节为20μl之后,进行聚合酶链式反应(polymerase chain reation),测定g13d突变(mutant)。此时,反应条件在95℃温度下初始变性10分钟之后,在95℃温度下10秒钟并在64℃温度下60秒钟,如此进行40个循环,实时测定每个循环的信号(hex),来导出结果。相关结果示于图2中。
[0142]
实施例2
‑
2:利用根据本发明的单核酸的实时kras基因的野生型分析
[0143]
以与上述实施例2
‑
1相同的条件,将序列号8的kras基因野生型单核酸和序列号9及10的引物中分别添加从nci
‑
h1975细胞株提取的总基因组dna各30ng、3ng、300pg、30pg和从hct
‑
15细胞株提取的总基因组dna各30ng、3ng、300pg、30pg之后,测定kras野生型基因。此时的结果示于图3中。hct
‑
15细胞株为以杂合型(heterozygous type)的基因同时保留g13d突变(参照图2的结果)和kras野生型的基因的细胞株,即使具有一半的野生型也可获
得如图4所示的结果。
[0144]
实施例2
‑
3:利用根据本发明的单核酸的与kras基因的野生型混合的g13d突变的实时分析
[0145]
待测定的kras基因在从培养规定时间的hct
‑
15细胞株和nci
‑
h1975细胞株提取的总基因组dna(total genomic dna)中进行检测。总基因组dna使用purelink genomic dna mini kit(美国赛默飞世尔科技公司(thermo fisher scientific),cat no.k1820
‑
00)在各个细胞株的5x106细胞中提取,并利用nanodrop one(美国赛默飞世尔科技公司(thermo fisher scientific))进行定量。定量的nci
‑
h1975 dna稀释为30ng/ul,hct
‑
15dna稀释为30ng/ul之后,按各1/10进行系列稀释(serial dilution),获得3ng/μl~3pg/μl浓度的nci
‑
h1975基因组dna及hct
‑
15基因组dna。之后,分别混合基因组dna各2μl,使得hct
‑
15的浓度为nci
‑
h1975浓度的10~0.01%,用于实验。据悉,hct
‑
15细胞株为g13d突变细胞株,nci
‑
h1975为kras野生型细胞株。
[0146]
然后,使用上述制备的序列号7的10um浓度的g13d单核酸0.3μl,序列号9及10的10um浓度的引物分别准备0.5μl。其中分别添加0.5u rnase
‑
h、aptataq dna master(瑞士罗氏(roche)公司制造)4μl、稀释为10~0.01%的dna之后,用三次蒸馏水调节总体积(total volume)为20μl,然后进行聚合酶链式反应(polymerase chain reation),测定g13d突变(mutant)。此时,反应条件在95℃温度下初始变性10分钟之后,在95℃温度下10秒钟并在64℃温度下60秒钟,如此进行40个循环,实时测定每个循环的信号(hex),来导出结果。相关结果示于图5中。
[0147]
如图2至图5所示,利用g13d单核酸(1型)进行g13d突变检测反应时,不引起与nci
‑
h1975野生型基因组dna之间的交叉反应,因此特异性优异,但在hct
‑
15基因组dna 600pg以下时检测不准确,这在包含野生型和点突变(point mutation)基因的分析的情况下确认到难以进行点突变(point mutation)基因小于10%的分析。
[0148]
实施例3:利用2型单核酸的apoe分析
[0149]
为了测定对于apoe基因的密码子(codon)112及密码子158的等位基因是否出现snp,使用了根据本发明的2型单核酸。如下表3所示,委托美国集成dna技术公司(integrated dna technologies;idt,usa))制备了2型单核酸。
[0150]
其中,就2型单核酸而言,作为具有x
‑
y
‑
z结构的引物及探针,在5'
‑
末端分别附着fam、hex、texasred,并且在各个3'
‑
末端附着iabkfq。并且,为了区别核糖核苷酸(rna)和脱氧核糖核苷酸(dna),在核糖核苷酸(rna)的序列前用角注“r”表示。
[0151]
表3
[0152]
4种apoe单核酸(2型)
[0153]
[0154]
为了分析,从韩国细胞株库分发了人细胞株pc3(e2/e2)、a549(e3/e3)、u937(e4/e4),从中获得了基因组dna。为了分析6种表型,同型表型使用了pc3(e2/e2)、a549(e3/e3)、u937(e4/e4),异型表型以高浓度在每次反应中包含32ng(约104拷贝)的方式将作为各个基因组dna的混合型的pc3 a549(e2/e3)、pc3 u937(e2/e4)、a549 u937(e3/e4)使用于分析。
[0155]
上述表3的apoe单核酸1、2、3、4的0.375μm、0.1μm、0.25μm及0.25μm的各个最终浓度下,使用上述基因组dna和0.1ng的耐热性rnase h以及aptataq dna master w/o mgcl2(瑞士罗氏(roche)公司制造)4μl、gc rich solution(瑞士罗氏(roche)公司制造)4μl、2.75mm的mgcl2、62.5nm的低rox(low rox)、无核酸酶的水(nuclease
‑
free water)将总体积调节为20μl之后,进行聚合酶链式反应(pcr)。此时,pcr反应条件在95℃温度下10分钟,在95℃温度下15秒钟,在64℃温度下55秒钟,如此进行40个循环。相关结果示于图6中。
[0156]
其结果,在利用根据本发明的2型单核酸的apoe的各等位基因的组合6种的分析中,对于密码子112的586t突变和密码子158的724t突变而言,具有可通过相同荧光染料的终点(end point)荧光值的差异区分而不是通过荧光染料差异的分析的局限性。由于这种局限性,根据各个dna样品的浓度读取结果存在一些问题。这表明,对于先天突变的apoe而言,如实施例1所示,利用1型单核酸相比于利用2型单核酸更易于分析。
[0157]
实施例4:利用2型单核酸的kras突变分析
[0158]
实施例4
‑
1:利用根据本发明的2型单核酸的g12v、g12c、g12s突变实时分析
[0159]
利用2型单核酸来测定了kras基因是否产生12密码子突变3种(g12v、g12c、g12s)的突变(mutant)。委托idt,如下表4制备了根据本发明的单核酸和uni
‑
反向引物(uni
‑
reverse primer)。就单核酸而言,在5'
‑
末端附着hex、fam、cy5,并且在3'
‑
末端附着iabkfq。并且,为了区别核糖核苷酸(rna)和脱氧核糖核苷酸(dna),在核糖核苷酸(rna)的序列前用角注“r”表示。
[0160]
表4
[0161]
利用于kras突变分析的单核酸及引物
[0162][0163]
为了利用上述合成的单核酸来确认kras点突变(point mutation)检测能力,分别培养野生型细胞株及突变细胞株,总基因组dna利用purelink基因组dna迷你试剂盒(purelink genomic dna mini kit,美国赛默飞世尔科技公司(thermo fisher scientific),cat no.k1820
‑
00)从各个细胞株的5
×
106细胞中提取。另一方面,利用nanodrop one(美国赛默飞世尔科技公司(thermo fisher scientific))定量之后用作模板(template)。使用的细胞株为如下表5所示。
[0164]
表5
[0165]
kras基因的12密码子突变3种(g12v、g12c、g12s)的突变细胞株
[0166][0167]
然后,上述制备的序列号15的g12v单核酸和序列号18的引物使用了10um浓度0.5μl,向5
×
aptataq dna master(瑞士罗氏(roche)公司制造)3.6μl和耐热性rnase h 0.2μl中添加colo201(kras野生型细胞株)30ng和从sw620(g12v突变)细胞株提取的总基因组dna各3ng、300pg、30pg、3pg之后,用三次蒸馏水将总体积调节为20μl之后,进行聚合酶链式反应(pcr),测定g12v突变。此时,反应条件在95℃温度下初始变性10分钟之后,在95℃温度下15秒钟并在66℃温度下30秒钟,如此进行4个循环,进行第一次pcr反应之后,在85℃温度下15秒钟并在64℃温度下40秒钟,如此进行40个循环,进行第二次pcr反应,实时测定每个循环的信号(hex),来导出结果。相关结果示于图7中。
[0168]
并且,上述制备的序列号16的g12c单核酸和序列号18的引物使用10um浓度0.5μl,向5x aptataq dna master(瑞士罗氏(roche)公司制造)2.8μl和5x apta fast buffer 1.2μl、1u/μl的耐热性rnase h 0.4μl、25mm mgcl
2 0.5μl中分别添加colo201(kras野生型细胞株)70ng和从mia
‑
paca2(g12c突变)细胞株提取的总基因组dna各7ng、700pg、70pg、7pg,用三次蒸馏水将总体积调节为20μl之后,进行聚合酶链式反应,测定g12c突变。此时,反应条件在95℃温度下初始变性10分钟之后,在95℃温度下10秒钟并在64℃温度下60秒钟,如此进行40个循环,每个循环实时测定信号(cy5),来导出结果。相关结果示于图8中。
[0169]
并且,使用上述制备的序列号17的g12s单核酸和序列号18的引物10um浓度0.5μl,向5x aptataq dna master(瑞士罗氏(roche)公司制造)3.6μl和10ng/μl的耐热性rnase h 0.2μl中分别添加colo201(kras野生型细胞株)30ng和从a549(g12s突变)细胞株提取的总基因组dna各3ng、300pg、30pg、7pg,用三次蒸馏水将总体积调节为20μl之后,进行聚合酶链式反应(polymerase chain reation),测定g12s突变。此时,反应条件在95℃温度下初始变性10分钟之后,在95℃温度下10秒钟并在64℃温度下60秒钟,如此进行40个循环,每个循环实时测定信号(fam),来导出结果。相关结果示于图9中。
[0170]
如上述图7至图9所示,根据本发明的单核酸在突变(point mutation)基因小于0.01%的情况下,能够以优异的特异性及灵敏度分析突变基因。
[0171]
实施例5:利用2型单核酸的egfr突变分析方法
[0172]
表皮生长因子受体(epidermal growth factor receptor;egfr)在非小细胞肺癌中超表达,成为egfr酪氨酸激酶(tyrosine kinase;tki)的靶标。通过分析对应于该egfr基因的外显子(exon)18、19、20、21的酪氨酸激酶结构域(tyrosine kinase domain)中产生的突变,从而可预测对非小细胞癌患者的治疗剂的药物反应性,因此突变分析有助于患者的药物处方及治疗。分析其中使用频率最高的t790m(c2369t)突变体是否产生突变。
[0173]
如下表6所示,将根据本发明的2型单核酸及引物(primer)委托于美国集成dna技术公司(integrated dna technologies;idt,usa))制备。其中,就t790m单核酸而言,在5'
‑
末端附着fam,并且在3'
‑
末端附着iabkfq。另一方面,为了区别核糖核苷酸(rna)和脱氧核
糖核苷酸(dna),在核糖核苷酸(rna)的序列前用角注“r”表示。
[0174]
表6
[0175]
利用egfr突变分析的单核酸及引物
[0176][0177]
为了分析,从作为t790m突变细胞株的h1975和作为野生型细胞株的a549中获得了基因组dna。h1975及a549基因组dna以30ng(约1
×
104拷贝)定量之后,分别稀释10倍来使用。
[0178]
在上述表6的t790m单核酸0.25μm、t790m引物以0.25μm的最终浓度存在之下,在上述基因组dna、0.5unit的耐热性rnase h、5
×
aptataq dna master(瑞士罗氏(roche)公司制造)3.6μl中,用无核酸酶的水(nuclease
‑
free water)调节总体积为20μl之后,进行聚合酶链式反应(pcr)。此时,pcr反应条件在95℃温度下10分钟,在95℃温度下15秒钟,在64℃温度下60秒钟,如此进行45个循环。相关结果示于图10中。
[0179]
其结果,在利用根据本发明的单核酸的egfr突变分析中,分别比较各个突变型和野生型时,确认到可区分至0.1%以下。如上所述,在如癌症(cancer)的点突变(point mutation)等的表观遗传突变的情况下,利用2型单核酸来进行探测是有利的。
[0180]
实施例6:利用2型单核酸的let
‑
7mirna及mirna 34异构体分析
[0181]
实施例6
‑
1:利用根据本发明的2型单核酸的let
‑
7mirna实时分析
[0182]
据悉,let
‑
7mirna在mirna中存在最多的异构体(isoform)。已知,区别这种let
‑
7的异构体是非常困难的分析,一般小于1%的特异性区别非常困难。为了测定本实验在其中let
‑
7a(5'
‑
ugagguaguagguuguauaguu)和let
‑
7d(5'
‑
agagguaguagguugcauaguu)的基因是否表达,将根据本发明的2型单核酸和引物(primer)、rt
‑
引物委托美国集成dna技术公司(integrated dna technologies;idt,usa)),如下表7制备。并且,为了准确的定量,委托idt制备了上述let
‑
7的mirna。其中,就单核酸而言,在5'
‑
末端附着荧光素琥珀酰亚胺酯(fluorescein succinimidyl ester;fam),并且在3'
‑
末端附着3iabkfg。另一方面,为了区分核糖核苷酸(rna)和脱氧核糖核苷酸(dna),在核糖核苷酸(rna)的序列前用角注“r”表示。
[0183]
表7
[0184]
用于区别let
‑
7mirna异构体的单核酸及引物
[0185][0186]
利用合成的各20pm浓度的mirna 1μl和美国ambion公司的聚a尾试剂盒(poly
‑
a tailing kit)及瑞士罗氏(roche)公司的nxtscript rt kit(总20μl),在45℃温度下,10μm浓度的1μl的上述表7的各个rt引物存在之下反应30分钟,合成cdna。
[0187]
将上述合成的cdna浓度从100fm稀释至1am。
[0188]
上述表7的单核酸及引物分别以10μm浓度0.5μl存在之下,加入上述合成后稀释的各个cdna 2μl、1u的耐热性rnase h、aptataq dna master(瑞士罗氏(roche)公司制备)4μl,用三次蒸馏水将总体积调节为20μl之后,进行聚合酶链式反应(pcr)。此时,pcr反应条件在95℃温度下5分钟,在63~64℃温度下60秒钟,在95℃温度下10秒钟,如此进行45个循环。相关结果示于图11及图12中。
[0189]
其结果,确认到仅使用mirna 1am浓度的稀释cdna(约1拷贝)2μl就可分析每个mirna。
[0190]
实施例6
‑
2:利用根据本发明的2型单核酸的let
‑
7mirna特异性实时检测
[0191]
在上述表7的单核酸及引物分别以10μm浓度存在0.5μl时,1pm浓度的let
‑
7d cdna 2μl中,按各1/10添加将浓度从100fm稀释至1am的let
‑
7a cdna 2μl、1u的耐热性rnase h、aptataq dna master(瑞士罗氏(roche)公司制备)4μl,用三次蒸馏水将总体积调节为20μl之后,进行聚合酶链式反应(pcr)。此时,pcr反应条件在95℃温度下5分钟,在63~64℃温度下60秒钟,在95℃温度下10秒钟,如此进行45个循环。相关结果示于图13中。
[0192]
其结果,在实验组中,向1pm浓度的let
‑
7d cdna 2μl中,按各1/10添加将浓度从100fm稀释至1am的let
‑
7a cdna各2μl,由此确认到从100fm至1fm浓度可稳定分析。这表明,可将异构体(isoform)的mirna特异性保持至0.1%来进行分析。
[0193]
实施例6
‑
3:利用根据本发明的2型单核酸的mirna 34a、mirna 34b及mirna 34c实时分析
[0194]
据悉,mirna 34存在3种异构体(isoform),区别这些异构体相当困难。在本实施例中,为了测定mirna 34a、mirna 34b及mirna 34c的基因是否表达,委托idt,如表8所示合成及制备了单核酸和引物、rt
‑
引物。就单核酸而言,在5'
‑
末端附着荧光素琥珀酰亚胺酯(fluorescein succinimidyl ester;fam)及六氯荧光素(hexachloro
‑
fluorescein;hex),并且在3'
‑
末端附着3iabkfq。并且,为了区分核糖核苷酸(rna)和脱氧核糖核苷酸(dna),核糖核苷酸(rna)用角注“r”表示。
[0195]
表8
[0196]
用于分析mirna 34a、mirna 34b及mirna 34c的单核酸及引物
[0197][0198][0199]
利用美国英杰(invitrogen)公司的聚a尾试剂盒(poly
‑
a tailing kit)和瑞士罗氏(roche)公司的nxtscript rt kit(总20μl),每个mirna在40℃温度的10μm浓度的rt引物1μl存在之下,反应60分钟,合成cdna,将合成的cdna从1pm至1am的浓度(约106~100拷贝)按各1/10稀释7个浓度,进行聚合酶链式反应(pcr)。在pcr中,加入各个合成cdna、耐热性rnase h、aptataq dna master(瑞士罗氏(roche)公司)、单核酸、引物,用三次蒸馏水将总体积调节为20μl之后,反应条件在95℃温度下5分钟,在95℃温度下10秒钟,在65℃温度下1分钟,如此进行45个循环。
[0200]
其结果,确认到mirna 34a、mirna 34b及mirna 34c均可从1pm至10am浓度(约106~101拷贝)以正常pcr效率进行测定。
[0201]
相关结果示于图14至图16中。
[0202]
实施例6
‑
4:利用根据本发明的2型单核酸的mirna 34a、mirna 34b及mirna 34c特异性实时检测
[0203]
在上述实施例6
‑
3中确认到mirna 34a、mirna 34b及mirna 34c单核酸可区分各个异构体(isoform),在本实施例中,确认了待扩增的mirna与其他异构体mirna混合时可区分异构体的比率程度。向108(100pm)mirna34a或mirna 34b异构体中混合了按各1/10稀释107至104或103(10pm~10fm或10pm~1fm)浓度的mirna 34c,利用mir
‑
34c单核酸和引物,通过聚合酶链式反应(pcr)进行特异性检测。
[0204]
在pcr中,加入各个合成cdna、耐热性rnase h、aptataq dna master(瑞士罗氏(roche)公司制备)、单核酸、引物,用三次蒸馏水将总体积调节为20μl,反应条件在95℃温度下5分钟,在95℃温度下10秒钟,在65℃温度下1分钟,如此进行45个循环。
[0205]
相关结果示于图17及图18中。
[0206]
其结果,确认到mirna 34c将异构体特异性保留至最大0.001%,并且可区分。
[0207]
实施例7:根据单核酸结构的分析方法
[0208]
为了在检测根据本发明的单核酸(2型)的突变时提高特异性,将g12d的3种类型委托于美国集成dna技术公司(integrated dna technologies;idt,usa))制备,具体地,为了
了解根据单核酸的核糖核苷酸(rna)的位置的突变(mutant)检测能力,委托美国集成dna技术公司(integrated dna technologies;idt,usa))如下表9制备了r1drmr2、r1rdmr2、r1dmrdr2类型。在各个单核酸的5'
‑
末端附着fam,并且在3'
‑
末端附着3iabkfq。并且,为了区别核糖核苷酸(rna)和脱氧核糖核苷酸(dna),在核糖核苷酸(rna)的序列前用角注“r”表示。
[0209]
表9
[0210]
对于kras基因g12d的3种类型(r1drmr2、r1rdmr2、r1dmrdr2)的单核酸
[0211][0212]
*r1及r2:dna寡聚(dnaoligo),d:dna,r:rna,m:突变(mutation)
[0213]
实施例7
‑
1:根据g12d单核酸类型(g12d
‑
r1drmr2、g12d
‑
r1rdmr2、g12d
‑
r1dmrdr2)的g12d突变基因和kras野生型基因分析
[0214]
准备10μm浓度的单核酸序列号36至38和引物序列号18各0.5μl,添加0.5u rnase h、aptataq master(瑞士罗氏(roche)公司制造)3.6μl、从aspc
‑
1(g12d突变型)及ht
‑
29(kras野生型)提取的总基因组dna30ng之后,用三次蒸馏水将总体积调节为20μl之后,进行聚合酶链式反应(pcr)。此时,反应条件在95℃温度下初始变性10分钟之后,在95℃温度下10秒钟,在64℃温度下60秒钟,如此反应45个循环,每个循环实时测定信号(fam),来导出结果。相关结果示于图19至图21中。
[0215]
根据单核酸的三种结构,确认到在kras野生型基因的特异性上示出其他结果,三种结构均可区分点突变(point mutation)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
