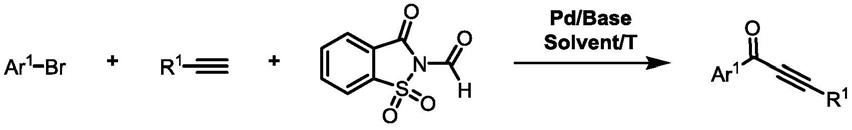
1.本发明属于有机合成技术领域,尤其涉及一种炔酮类化合物的制备方法。
背景技术:
2.炔酮(ynone)具有特殊共轭结构单元,是一类用途非常广泛的有机合成中间体。目前炔酮的合成方法包括:(1)炔基金属试剂与酰氯/酸酐等的反应;(2)钯催化末端炔烃与卤代烃的插羰基反应;(3)炔醇的氧化反应;(4)非端炔的亚甲基氧化反应等。其中,钯催化末端炔烃与卤代烃的插羰基反应由于原料易得,反应高效,适用范围广等特点,具有极高的实用价值。
3.传统钯催化末端炔烃与卤代烃的插羰基反应中主要选用碘代芳烃作为原料,作为反应原料碘苯的价格远高于溴苯(以国产试剂平台安耐吉为例:碘代苯¥508.00元/0.5kg,溴代苯¥98元/0.5kg),因此溴代芳烃代替碘代芳烃参与制备炔酮的反应具有重要的实用价值。然而使用的剧毒、易爆的一氧化碳(co)、具有强烈刺激气味的异氰等作为羰基来源严重限制了此类反应的应用,因此开发一种安全高效插羰基反应合成炔酮具有非常重要的意义。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种炔酮类化合物的制备方法,本发明提供的方法廉价、适用范围宽泛、环境友好。
5.本发明提供了一种炔酮类化合物的制备方法,包括:
6.将式(ii)结构化合物、式(iii)结构化合物和n
‑
甲酰基糖精在催化剂和碱的作用下于非水溶剂中进行反应,得到式(i)结构的炔酮类化合物:
7.ar1‑
br
ꢀꢀ
式ii;
8.式ii和式i中,ar1选自芳基、杂芳基及其衍生物;
9.式iii和式i中,r1选自烷基,芳基,杂芳基,酰基及其衍生物。
10.优选的,所述ar1选自取代或未取代的c6~c20的芳基、取代或未取代的c6~c20的杂芳基。
11.优选的,所述r1选自h、取代或未取代的c1~c10的烷基、取代或未取代的c6~c20的芳基、取代或未取代的c6~c20的杂芳基、取代或未取代的c1~c10的酰基。
12.优选的,所述催化剂为钯类催化剂。
13.优选的,所述钯类催化剂选自醋酸钯、四三苯基膦钯和双三苯基膦二氯化钯中的一种或多种。
14.优选的,所述催化剂的摩尔量为式ii结构化合物摩尔量的0.5~100%。
15.优选的,所述碱选自碳酸钾、碳酸钠、氢氧化钾、三乙胺和二异丙基胺等常见有机
无机碱中的一种或多种。
16.优选的,所述式iii结构化合物的用量相对于式ii结构式化合物为1~5equiv。
17.优选的,所述n
‑
甲酰基糖精的用量相对于式ii结构化合物为1~5equiv。
18.优选的,所述非水溶性溶剂选自甲苯、苯、三氟甲苯、1,4
‑
二氧六环、乙腈、二甲基甲酰胺和二甲基亚砜中的一种或多种。
19.本发明提供了一种采用插羰基sonogashira反应合成炔酮新方法,本发明提供的方法选择安全高效的n
‑
甲酰基糖精,由溴代芳香烃出发通过插羰基反应合成炔酮,降低了反应的成本,拓宽了反应的适用范围,避免了具有危险、强烈刺激气味的羰基来源的使用。本发明提供的方法可以高效地得到高纯度的炔酮类化合物及其衍生物。
20.本发明中,n
‑
甲酰基糖精具有廉价易得、稳定安全、易于储存的优点,本发明提供的方法具有操作简单,原料和试剂易得,条件温和,催化体系绿色环保,产物易分离纯化等优点,适用于合成各种类型的炔酮类化合物,可以高效、高收率地制得高纯度的炔酮类化合物及其衍生物。
附图说明
21.图1为实施例1制备的炔酮类化合物的1h
‑
nmr核磁共振谱图;
22.图2为实施例1制备的炔酮类化合物的
13
c
‑
nmr核磁共振谱图;
23.图3为实施例2制备的炔酮类化合物的1h
‑
nmr核磁共振谱图;
24.图4为实施例2制备的炔酮类化合物的
13
c
‑
nmr核磁共振谱图。
具体实施方式
25.下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员经改进或润饰的所有其它实例,都属于本发明保护的范围。应理解,本发明实施例仅用于说明本发明的技术效果,而非用于限制本发明的保护范围。实施例中,所用方法如无特别说明,均为常规方法。
26.本发明提供了一种炔酮类化合物的制备方法,包括:
27.将式(ii)结构化合物、式(iii)结构化合物和n
‑
甲酰基糖精在催化剂和碱的作用下于非水溶剂中进行反应,得到式(i)结构的炔酮类化合物:
28.ar1‑
br
ꢀꢀ
式ii;
29.式ii和式i中,ar1选自芳基、杂芳基及其衍生物;
30.式iii和式i中,r1选自烷基,芳基,杂芳基,酰基及其衍生物。
31.本发明制备炔酮类化合物的过程中选用芳基溴代物作为反应底物。
32.在本发明中,所述式ii和式i中,ar1优选选自取代或未取代的c6~c20的芳基、取代或未取代的c6~c20的杂芳基,更优选选自苯基、对甲基苯基、对甲氧基苯基、对乙酰基苯基、萘基、噻吩基、吲哚基等。
33.在本发明中,所述式iii和式i中,r1优选选自h、取代或未取代的c1~c10的烷基、取代或未取代的c6~c20的芳基、取代或未取代的c6~c20的杂芳基、取代或未取代的c1~c10的酰基,更优选选自正己基、正戊基、苄基、苯基、对甲基苯基、对甲氧基苯基、对乙酰基苯基、萘基、噻吩基、吲哚基等。
34.在本发明中,所述式ii结构化合物优选选自溴苯、对溴甲苯、对溴苯基醚、2
‑
溴萘、2
‑
溴噻吩、5
‑
溴吲哚等。
35.在本发明中,所述式iii结构化合物优选选自正辛炔、正庚炔、苯乙炔、对甲基苯乙炔、2
‑
噻吩乙炔、3
‑
噻吩乙炔、2
‑
萘乙炔等。
36.在本发明中,所述n
‑
甲酰基糖精作为一氧化碳供体。
37.在本发明中,所述催化剂优选为钯类催化剂。
38.在本发明中,所述钯类催化剂选自醋酸钯、四三苯基膦钯和双三苯基膦二氯化钯中的一种或多种。
39.在本发明中,所述非水溶性溶剂选自甲苯、苯、三氟甲苯、1,4
‑
二氧六环、乙腈、二甲基甲酰胺和二甲基亚砜中的一种或多种。
40.在本发明中,相对于式(ii)结构化合物,式(iii)结构化合物的用量优选为1~5equiv,更优选为2~4equiv,更优选为2~3equiv,最优选为2equiv。
41.在本发明中,相对于式ii结构化合物,n
‑
甲酰基糖精的用量优选为1~5equiv,更优选为2~3equiv,最优选为3equiv。
42.在本发明中,所述催化剂的摩尔量优选为式ii结构化合物摩尔量的0.5~100%,更优选为5~80%,更优选为10~60%,更优选为20~50%,最优选为30~40%,最最优选为10%。
43.在本发明中,相对于式ii结构化合物,碱的用量优选为1~5equiv,更优选为2~4equiv,最优选为3equiv。
44.在本发明中,相对于式ii结构化合物,所述非水溶性溶剂的用量优选为1~10ml/mmol(即每mmol式ii结构化合物采用1~10ml的非水溶性溶剂),更优选为2~8ml/mmol,最优选为4ml/mmol。
45.在本发明中,所述反应优选在惰性气体的保护下进行,所述惰性气体优选为氮气。
46.在本发明中,所述反应的温度优选为80~160℃,更优选为100~140℃,更优选为100~120℃,最优选为100℃。在本发明中,所述反应过程中优选油浴加热。
47.在本发明中,所述反应过程中优选在搅拌的条件下进行,优选搅拌加热;所述反应过程中优选在密封的条件下进行。
48.在本发明中,所述反应过程中采用tlc检测底物是否消失,底物消失反应结束。
49.在本发明中,所述反应的方程式如下:
[0050][0051]
在本发明中,所述炔酮类化合物的制备方法优选包括:
[0052]
将式ii结构化合物溴代芳香烃、式iii结构化合物端炔类化合物和n
‑
甲酰基糖精
在钯(pd)催化和碱的作用下于非水溶剂中进行反应,得到式i结构化合物炔酮类化合物及其衍生物。
[0053]
在本发明中,所述反应结束后优选还包括:
[0054]
将得到的反应产物进行硅胶柱层析,得到炔酮类化合物。
[0055]
在本发明中,所述硅胶柱层析过程中采用石油醚和乙醚;所述石油醚和乙醚的体积比优选为100:(1~3),更优选为100:(1.5~2.5),最优选为100:2。
[0056]
本发明提供了一种采用插羰基sonogashira反应合成炔酮新方法,本发明提供的方法选择安全高效的n
‑
甲酰基糖精,由溴代芳香烃出发通过插羰基反应合成炔酮,降低了反应的成本,拓宽了反应的适用范围,避免了具有危险、强烈刺激气味的羰基来源的使用。本发明提供的方法可以高效地得到高纯度的炔酮类化合物及其衍生物。
[0057]
本发明中,n
‑
甲酰基糖精具有廉价易得、稳定安全、易于储存的优点,本发明提供的方法具有操作简单,原料和试剂易得,条件温和,催化体系绿色环保,产物易分离纯化等优点,适用于合成各种类型的炔酮类化合物,可以高效、高收率地制得高纯度的炔酮类化合物及其衍生物。
[0058]
实施例1合成炔酮4a
[0059]
按照下述方程式进行反应:
[0060][0061]
氮气保护下,依次将溴苯1a(0.16g,1mmol)、苯乙炔2a(0.21g,2mmol)、n
‑
甲酰基糖精3(0.42g,2mmol)、碳酸钾(0.55g,4mmol)加入甲苯中,然后加入醋酸钯(22mg,0.1mmol)加入,密封并放入100℃油浴中搅拌加热。tlc检测底物消失,反应结束,经过硅胶柱层析(v
石油醚
:v
乙醚
=100:2)得到白色固体0.18g,经过nmr检测(检测结果如图1和图2所示)、ms检测,证实实施例1提供的方法制备的炔酮化合物结构如4a所示,其收率以溴苯1a为基础为87%,纯度>99%。
[0062]
谱图解析数据如下:
[0063]1h nmr(500mhz,cdcl3)δ8.26
–
8.20(m,2h),7.72
–
7.68(m,2h),7.64(t,j=7.4hz,1h),7.53(t,j=7.8hz,2h),7.49(d,j=7.3hz,1h),7.43(t,j=7.4hz,2h);
[0064]
13
c nmr(126mhz,cdcl3)δ178.18,136.99,134.27,133.21,130.94,129.71,128.82,128.76,120.24,93.26,87.01;
[0065]
hrms(esi)m/z calculated for c
15
h
10
o[m h]
:207.1449,found 207.1450。
[0066]
实施例2
[0067]
按照下述方程式进行反应:
[0068]
[0069]
按照实施例1的方法制备炔酮化合物,与实施例1的区别在于,将1a化合物替换为1b化合物。
[0070]
经过nmr检测(检测结果如图3和图4所示)、ms检测,证实实施例2提供的方法制备的炔酮化合物结构如4b所示,其收率以1b为基础为91%,,纯度>99%。
[0071]
谱图解析数据如下:
[0072]1h nmr(500mhz,cdcl3)δ8.12(d,j=8.1hz,2h),7.68(d,j=7.1hz,2h),7.48(t,j=7.4hz,1h),7.42(t,j=7.4hz,2h),7.31(d,j=7.9hz,2h),2.45(s,3h).;
[0073]
13
c nmr(126mhz,cdcl3)δ177.86,145.38,134.67,133.14,130.81,129.82,129.69,129.46,128.77,120.32,92.75,87.06,21.98;
[0074]
hrms(esi)m/z calculated c
16
h
12
o[m h]
:221.0765,found 221.0757。
[0075]
实施例3
[0076]
按照下述方程式进行反应:
[0077][0078]
按照实施例1的方法制备炔酮化合物,与实施例1的区别在于,将1a化合物替换为1c化合物。
[0079]
经过nmr检测、ms检测,证实实施例3提供的方法制备的炔酮化合物结构如4c所示,其收率以1c为基础为90%,纯度>99%。
[0080]
谱图解析数据如下:
[0081]1h nmr(500mhz,cdcl3)δ8.20(d,j=8.4hz,2h),7.68(d,j=7.5hz,2h),7.47(d,j=7.0hz,1h),7.42(t,j=7.5hz,2h),6.99(d,j=8.5hz,2h),3.91(s,3h);
[0082]
13
c nmr(126mhz,cdcl3)δ176.68,164.44,132.94,131.98,130.57,130.25,128.62,120.31,113.85,92.31,86.87,77.25,77.00,76.75,55.60;
[0083]
hrms(esi)m/z calculated c
16
h
12
o2[m h]
:237.0863,found 237.0866。
[0084]
实施例4
[0085]
按照下述方程式进行反应:
[0086][0087]
按照实施例2的方法制备炔酮化合物,与实施例2的区别在于,将2a化合物替换为2b化合物。
[0088]
经过nmr检测、ms检测,证实实施例4提供的方法制备的炔酮化合物结构如4d所示,其收率以1b为基础为91%,纯度>99%。
[0089]
谱图解析数据如下:
[0090]1h nmr(500mhz,cdcl3)δ8.11(d,j=8.1hz,2h),7.57(d,j=8.0hz,2h),7.30(d,j=7.9hz,2h),7.21(d,j=7.9hz,2h),2.43(s,3h),2.39(s,3h);
[0091]
13
c nmr(126mhz,cdcl3)δ177.84,145.19,141.50,134.71,133.14,133.03,129.73,129.60,129.53,129.38,128.66,117.15,93.39,86.90,21.91,21.84;
[0092]
hrms(esi)m/z calculated c
17
h
14
o[m h]
:235.1065,found 235.1066。
[0093]
实施例5
[0094]
按照下述方程式进行反应:
[0095][0096]
按照实施例2的方法制备炔酮化合物,与实施例2的区别在于,将2a化合物替换为2c化合物。
[0097]
经过nmr检测、ms检测,证实实施例5提供的方法制备的炔酮化合物结构如4e所示,其收率以1b为基础为82%,纯度>99%。
[0098]
谱图解析数据如下:
[0099]1h nmr(500mhz,cdcl3)δ8.09(d,j=8.0hz,2h),7.61(d,j=8.3hz,2h),7.40(d,j=8.3hz,2h),7.31(d,j=8.0hz,2h),2.45(s,3h);
[0100]
13
c nmr(126mhz,cdcl3)δ177.65,145.58,137.19,134.56,134.34,133.81,129.85,129.53,129.26,129.02,118.84,91.27,87.79,22.02;
[0101]
hrms(esi)m/z calculated c
16
h
11
clo[m h]
:255.0465,found 255.0457。
[0102]
实施例6
[0103]
按照下述方程式进行反应:
[0104][0105]
按照实施例2的方法制备炔酮化合物,与实施例2的区别在于,将2a化合物替换为2d化合物。
[0106]
经过nmr检测、ms检测,证实实施例6提供的方法制备的炔酮化合物结构如4f所示,其收率以1b为基础为89%,纯度>99%。
[0107]
谱图解析数据如下:
[0108]1h nmr(500mhz,cdcl3)δ8.00(d,j=8.1hz,2h),7.24(d,j=8.0hz,2h),2.46(t,j=7.1hz,2h),2.39(s,3h),1.67
–
1.56(m,2h),1.47(dd,j=14.9,7.4hz,2h),0.93(t,j=7.3hz,3h);
[0109]
13
c nmr(126mhz,cdcl3)δ177.95,144.91,134.61,129.65,129.21,96.30,79.70,29.87,22.09,21.77,18.89,13.55;
[0110]
hrms(esi)m/z calculated c
14
h
16
o[m h]
:201.1201,found 201.1205。
[0111]
实施例7
[0112]
按照下述方程式进行反应:
[0113][0114]
按照实施例2的方法制备炔酮化合物,与实施例2的区别在于,将2a化合物替换为2e化合物。
[0115]
经过nmr检测、ms检测,证实实施例7提供的方法制备的炔酮化合物结构如4g所示,其收率以1b为基础为91%,纯度>99%。
[0116]
谱图解析数据如下:
[0117]1h nmr(500mhz,cdcl3)δ8.03(d,j=8.1hz,2h),7.28(d,j=8.0hz,2h),2.43(s,3h),0.31(s,9h);
[0118]
13
c nmr(126mhz,cdcl3)δ177.53,145.43,134.29,129.90,129.76,129.41,128.69,125.64,101.06,100.09,30.43,21.97,
‑
0.53;
[0119]
hrms(esi)m/z calculated c
13
h
16
osi[m h]
:217.0970,found 217.0972。
[0120]
本发明实施例所采用的反应原料结构式与制备得到的产物的结构式如下:
[0121]
[0122][0123]
由以上实施例可知,本发明提供了一种采用插羰基sonogashira反应合成炔酮新方法,本发明提供的方法选择安全高效的n
‑
甲酰基糖精,由溴代芳香烃出发通过插羰基反应合成炔酮,降低了反应的成本,拓宽了反应的适用范围,避免了具有危险、强烈刺激气味的羰基来源的使用。本发明提供的方法可以高效地得到高纯度的炔酮类化合物及其衍生物。
[0124]
本发明中,n
‑
甲酰基糖精具有廉价易得、稳定安全、易于储存的优点,本发明提供的方法具有操作简单,原料和试剂易得,条件温和,催化体系绿色环保,产物易分离纯化等优点,适用于合成各种类型的炔酮类化合物,可以高效、高收率地制得高纯度的炔酮类化合物及其衍生物。
[0125]
以上所述的仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。