1.本发明属于烯烃聚合催化剂的技术领域,特别是涉及一种吡啶胺基铪化合物及其制备方法和应用。
背景技术:
2.聚烯烃是一种应用广泛的材料。其具有易于加工使用、价格低廉等优点。烯烃聚合催化剂因具有调控聚烯烃结构和性能的能力,而成为聚烯烃工业的关键。因此,设计和开发新的聚烯烃催化剂成为推动聚烯烃工业发展的核心技术。
3.吡啶胺基铪催化剂首先由dow化学公司开发,显示出良好的催化烯烃聚合性能。吡啶胺基铪催化剂能够高活性催化乙烯聚合,制备得到线性的聚乙烯。它也能在高温下催化丙烯和其他α-烯烃聚合,制备得到等规的聚丙烯,表现出高活性、聚合物高分子量以及立构选择性等特点。同时它显示出良好的共聚性能,能够催化乙烯和α-烯烃共聚,制备得到高α-烯烃插入率聚烯烃的弹性体。特别是,它也能够作为链穿梭催化剂之一,和锆基催化剂一起催化乙烯和辛烯共聚,制备得到聚烯烃嵌段共聚物。dow化学公司通过链穿梭聚合技术,采用吡啶胺基铪催化剂已经成功的制备出了商业化的高性能聚烯烃嵌段共聚物(obc)。因而吡啶胺基铪催化剂在聚烯烃领域引起了广泛的重视。
4.吡啶胺基铪催化剂的结构对其催化性能有着重要的影响。除了骨架上的取代基能够影响其活性,分子量以及聚合物规整度外,芳胺邻位取代基也会对其聚合性能有着重要的影响。研究表明,芳胺邻位引入大位阻的取代基将进一步提高催化剂的热稳定性,同时也能够增强催化活性以及对α-烯烃聚合的立构选择性。因而当前的吡啶胺基铪催化剂的芳胺邻位一般都是异丙基取代。coates教授课题组报道了叔丁基取代的吡啶胺基铪催化剂,催化丙烯聚合,显示出了更好的活性,得到更高的分子量聚丙烯,并且聚丙烯的等规度明显提高。当前的芳胺邻位的取代基主要都是脂肪的烷基,还没有芳基的取代催化剂。
技术实现要素:
5.现有的吡啶胺基铪化合物邻位取代基都是异丙基,没有更大的取代基的化合物。如果刚性的芳环取代,一方面,芳环能够显著提高催化剂的热稳定性,也可以提高催化烯烃聚合产物分子量。同时由于大位阻也可以抑制链转移的发生,得到窄分布的聚合物。另一方面,更大的位阻取代基能够与增长链以及单体之间发生空间排斥作用,控制α-烯烃聚合的立构规整性,得到更高规整度的聚合物。另外,苯胺的对位现在还没有不同电子效应的取代基。而通过苯胺对位的电子效应,可以显著的调控烯烃聚合。
6.为此,本发明提供了一种新型芳胺邻位含有二苯甲基对位含有不同电子效应取代基的吡啶胺基铪化合物及其制备方法和应用。
7.这类吡啶胺基铪化合物具有以下结构:
[0008][0009]
所述式i中,r表示氢、甲基,x表示甲基、甲氧基、氟、氯、溴元素,可以调控电子效应。该化合物在助催化剂作用下,能够进行乙烯和α-烯烃的均聚与共聚。助催化剂为含硼化合物,或烷基铝与含硼化合物的混合物。
[0010]
本发明的第一个目的在于提供一种吡啶胺基铪化合物。
[0011]
本发明的第二个目的在于提供一种吡啶胺基铪化合物的制备方法。
[0012]
本发明的第三个目的在于提供一种吡啶胺基铪化合物在催化烯烃聚合方面的应用。
[0013]
为达到上述目的,本发明采用下述技术方案:
[0014]
一种吡啶胺基铪化合物,结构式如下:
[0015][0016]
所述式i中,r表示氢、甲基,x表示甲基、甲氧基、氟、氯、溴元素。
[0017]
上述的吡啶胺基铪化合物的制备方法,包括以下操作步骤:
[0018]
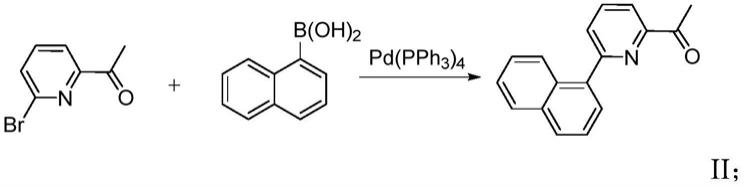
1)吡啶酮化合物与萘硼酸进行偶联反应,得到芳基取代的2-萘基-吡啶酮化合物。
[0019][0020]
2)将4-位取代的苯胺与二苯甲醇反应,得到取代的苯胺化合物。
[0021][0022]
3)2-芳基-吡啶酮化合物与取代的苯胺进行缩合反应,得到取代的吡啶亚胺化合
物。
[0023][0024]
4)将取代的吡啶亚胺化合物与强还原剂进行还原反应,制备得到取代的吡啶胺基化合物配体。
[0025][0026]
5)吡啶胺基化合物与强碱发生脱质子反应,然后加入四氯化铪的金属盐,制备得到相应的吡啶胺基铪氯化合物。吡啶胺基铪氯化合物继续与甲基溴化镁格式试剂发生甲基化反应,制备得到相应的吡啶胺基铪甲基化合物。
[0027][0028]
本发明还提供了这种新型铪化合物在助催化剂活化下在催化烯烃聚合方面的应用。
[0029]
所述的助催化剂为含硼化合物,或烷基铝与含硼化合物的混合物。含硼化合物组催化剂可以包括三乙基铵四(苯基)硼,三丁基铵四(苯基)硼、三甲基铵四(苯基)硼、三丙基铵(苯基)硼、三甲基铵四(对家苯基)硼、三甲基铵四(邻,对-二甲基苯基)硼、三丁基铵四(对三氟甲基苯基)硼、三甲基铵四(对三氟甲基苯基)硼、三丁基铵四(五氟苯基)硼、n,n-二乙基苯胺四(苯基)硼、n,n-二乙基苯胺四(五氟苯基)硼、二乙基苯胺四(五氟苯基)硼、三甲
基膦四(苯基)硼、三丙基铵四(对甲苯基)硼、三乙基铵四(对三氟甲基苯基)硼、三乙基铵四(邻,对-二甲基苯基)硼、三甲基铵四(邻,对-二甲基苯基)硼、三丁基铵四(对三氟甲基苯基)硼、三甲基铵四(对三氟甲基苯基)硼、三丁基铵四(五氟苯基)硼、三苯基膦四(苯基)硼、三(五氟苯)硼烷、三苯碳鎓四(五氟苯)硼、三苯碳鎓四(对三氟甲基苯基)硼、n,n-二甲基苯胺四(五氟苯基)硼、及三苯碳鎓四(五氟苯)硼酸盐或n,n-二甲基苯胺四(五氟苯基)硼酸盐等。
[0030]
所述烷基铝可包括三甲基铝、三乙基铝、三异丁基铝、三丙基铝、三丁基铝、二甲基氯化铝、三异丙基铝、三仲丁基铝、三戊基铝、三异戊基铝、三环戊基铝、三己基铝、三辛基铝、乙基二甲基铝、甲基二乙基铝、三苯基铝、三对甲基苯基铝、二甲基甲氧基铝等。所述助催化剂优选三(五氟苯)硼烷或三苯碳鎓四(五氟苯)硼酸盐或n,n-二甲基苯胺四(五氟苯基)硼酸盐或三异丁基铝和这几种含硼化合物的组合物,在该助催化剂作用下,能够高活性的催化乙烯和α-烯烃的均聚与共聚。
[0031]
所述主催化剂与助催化剂及烷基铝的hf:b:al比例为1:1.0~5.0:100~500(摩尔比)。
[0032]
所述的烯烃单体为乙烯、丙烯,己烯、辛烯或者两种及以上单体的混合物。
[0033]
与现有技术相比,本发明具有以下有益效果:
[0034]
(1)本发明在吡啶胺基铪化合物芳胺的邻位上引入大位阻刚性的二苯甲基,能够提高催化剂的热稳定性,同时提高了催化烯烃聚合的活性以及聚合物的分子量。
[0035]
(2)本发明大位阻刚性的二苯甲基在聚合过程中,能够与增长链以及单体之间发生空间排斥作用,能够控制α-烯烃聚合过程中的立构选择性,得到高等规度的聚合产物。
[0036]
(3)采用本发明的铪化合物催化烯烃聚合时,大的位阻能够抑制链转移链的发生,所得到的聚烯烃材料分子量分布窄。
[0037]
(4)本发明的铪化合物苯胺对位能够容易引入不同电子效应的取代基,调控催化剂的电子效应,进而更易于调控催化烯烃聚合的性能。
附图说明
[0038]
图1为实施例18制得的吡啶胺基铪化合物的核磁氢谱图。
[0039]
图2为实施例18制得的吡啶胺基铪化合物的核磁碳谱图。
[0040]
图3为实施例24制得的聚乙烯的dsc图。
[0041]
图4为实施例44制得的乙烯-辛烯共聚物的dsc图。
具体实施方式
[0042]
以下对本发明的实施例作详细说明:本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例,下列实施例中未注明具体条件的实验方法,通常按照常规条件。
[0043]
为了在实施例中清楚的表示配体和配合物,说明如下:
[0044]
n1为式iii中所示的取代苯胺,其中x为甲基;
[0045]
n2为式iii中所示的取代苯胺,其中x为甲氧基;
[0046]
n3为式iii中所示的取代苯胺,其中x为氟;
[0047]
n4为式iii中所示的取代苯胺,其中x为氯;
[0048]
n5为式iii中所示的取代苯胺,其中x为溴;
[0049]
b1为式iv中所示的吡啶亚胺化合物,其中r为氢,x为甲基;
[0050]
b2为式iv中所示的吡啶亚胺化合物,其中r为氢,x为甲氧基;
[0051]
b3为式iv中所示的吡啶亚胺化合物,其中r为氢,x为氟;
[0052]
b4为式iv中所示的吡啶亚胺化合物,其中r为氢,x为氯;
[0053]
b5为式iv中所示的吡啶亚胺化合物,其中r为氢,x为溴;
[0054]
b6为式iv中所示的吡啶亚胺化合物,其中r为甲基,x为甲基;
[0055]
配体l1为式v中所示的吡啶胺基配体,其中r为氢,x为甲基;
[0056]
配体l2为式v中所示的吡啶胺基配体,其中r为氢,x为甲氧基;
[0057]
配体l3为式v中所示的吡啶胺基配体,其中r为氢,x为氟;
[0058]
配体l4为式v中所示的吡啶胺基配体,其中r为氢,x为氯;
[0059]
配体l5为式v中所示的吡啶胺基配体,其中r为氢,x为溴;
[0060]
配体l6为式v中所示的吡啶胺基配体,其中r为甲基,x为甲基;
[0061]
配合物c1为式i中所示的吡啶胺基铪配合物,其中r为氢,x为甲基;
[0062]
配合物c2为式i中所示的吡啶胺基铪配合物,其中r为氢,x为甲氧基;
[0063]
配合物c3为式i中所示的吡啶胺基铪配合物,其中r为氢,x为氟;
[0064]
配合物c4为式i中所示的吡啶胺基铪配合物,其中r为氢,x为氯;
[0065]
配合物c5为式i中所示的吡啶胺基铪配合物,其中r为氢,x为溴;
[0066]
配合物c6为式i中所示的吡啶胺基铪配合物,其中r为甲基,x为甲基;
[0067]
一、配体的制备
[0068]
实施例1
[0069]
吡啶酮化合物的合成
[0070]
氮气氛围下,在支口瓶中依次加入2-乙酰基-6-溴吡啶1.86g(10mmol)、萘硼酸1.72g(10mmol)、四(三苯基磷)钯15mg、碳酸钾6g,打入30ml乙醇、20ml甲苯、10ml水,回流过夜。分液萃取,水洗、干燥,得到吡啶醛化合物a为2.17g,产率93%。核磁氢谱如下:1h nmr(300mhz,c6d6):δ8.19-8.15(m,1h,arh),8.01(dd,2.0hz,1h,arh),7.71-7.66(m,2h,arh),7.45(dd,1h,arh),7.33-7.28(m,3h,arh),7.18-7.13(m,2h,arh),2.59(s,3h,(ch3)co).
[0071]
实施例2
[0072]
取代苯胺n1的合成
[0073]
将二苯甲醇13.3g(72.2mmol)和4-甲基苯胺3.66克(34mmol)加入瓶中,加热至80℃熔化,加入盐酸溶解的氯化锌2.4g(17.6mmol),升温至150℃,反应2h后降温,用二氯甲烷溶解,碳酸氢钠中和多余的酸,分液萃取,旋干,用乙醇淋洗,得到白色粉末13.3g,产率89%。核磁氢谱如下:1h nmr(400mhz,cdcl3):δ7.34-7.27(d,8h,ph),7.24-7.18(d,4h,ph),7.12-7.06(d,8h,ph),6.38(s,2h,ar-h),5.45(s,2h,chph2),3.27(s,2h,nh2),2.02(s,3h,me).
[0074]
实施例3
[0075]
取代苯胺n2的合成
[0076]
按照实施例2中的合成方法,将实施例2中的4-甲基苯胺替换为4-甲氧基苯胺,产
率67%。核磁氢谱如下:1h nmr(400mhz,cdcl3):δ3.24(2h,bs,nh2),3.55(3h,s,och3),5.64(2h,s,chph2),6.40(2h,s,ar-h),7.25-7.43(20h,m,ar-h).
[0077]
实施例4
[0078]
取代苯胺n3的合成
[0079]
按照实施例2中的合成方法,将实施例2中的4-甲基苯胺替换为4-氟基苯胺,产率48%。核磁氢谱如下:1h nmr(400mhz,cdcl3):δ7.44-7.37(d,8h,ph),7.24-7.15(d,4h,ph),7.12-7.06(d,8h,ph),6.42(s,2h,ar),5.44(s,2h,chph2),3.26(s,2h,nh2).
[0080]
实施例5
[0081]
取代苯胺n4的合成
[0082]
按照实施例2中的合成方法,将实施例2中的4-甲基苯胺替换为4-氯基苯胺,产率71%。核磁氢谱如下:1h nmr(400mhz,cdcl3):δ7.44-7.37(d,8h,ph),7.24-7.15(d,4h,ph),7.12-7.06(d,8h,ph),6.42(s,2h,ar),5.44(s,2h,chph2),3.26(s,2h,nh2).
[0083]
实施例6
[0084]
取代苯胺n5的合成
[0085]
按照实施例2中的合成方法,将实施例2中的4-甲基苯胺替换为4-溴基苯胺,产率32%。核磁氢谱如下:1h nmr(400mhz,cdcl3):δ7.35-7.28(d,8h,ph),7.24-7.17(d,4h,ph),7.12-7.07(d,8h,ph),6.37(s,2h,ar),5.43(s,2h,chph2),3.25(s,2h,nh2).
[0086]
实施例7
[0087]
吡啶亚胺化合物b1的合成
[0088]
氮气氛围下,将吡啶酮化合物a2.33g(10mmol)、苯胺n1 4.62g(10.5mmol)、对甲苯磺酸10mg溶于50ml甲苯中,分水回流48h。旋干溶剂,干燥,得到吡啶亚胺化合物b1(6.53g),产率94%。核磁氢谱如下:1h nmr(cdcl3,400mhz):δ8.51(s,1h,nap-h),8.24-8.17(d,1h,py-h),8.01-7.78(m,6h,nap-h),7.57-7.47(m,2h,py-h),7.25-7.01(m,20h,ar-h),6.71(s,2h,ar-h),5.33(s,2h,ch),2.19(s,3h,ar-ch3),1.21(s,3h,c=n-ch3).
[0089]
实施例8
[0090]
吡啶亚胺化合物b2的合成
[0091]
按照实施例7中的合成方法,将实施例7中的n1替换为n2,产率81%。核磁氢谱如下:1h nmr(cdcl3,400mhz):δ8.52(s,1h,nap-h),8.25-8.17(d,1h,py-h),8.02-7.77(m,6h,nap-h),7.57-7.47(m,2h,py-h),7.25-7.01(m,20h,ar-h),6.71(s,2h,ar-h),5.33(s,2h,ch),2.23(s,3h,ar-och3),1.23(s,3h,c=n-ch3).
[0092]
实施例9
[0093]
吡啶亚胺化合物b3的合成
[0094]
按照实施例7中的合成方法,将实施例7中的n1替换为n3,产率74%。核磁氢谱如下:1h nmr(cdcl3,400mhz):δ8.51(s,1h,nap-h),8.23-8.17(d,1h,py-h),8.01-7.76(m,6h,nap-h),7.57-7.47(m,2h,py-h),7.25-7.01(m,20h,ar-h),6.72(s,2h,ar-h),5.31(s,2h,ch),1.23(s,3h,c=n-ch3).
[0095]
实施例10
[0096]
吡啶亚胺化合物b4的合成
[0097]
按照实施例7中的合成方法,将实施例7中的n1替换为n4,产率65%。核磁氢谱如
下:1h nmr(cdcl3,400mhz):δ8.52(s,1h,nap-h),8.25-8.17(d,1h,py-h),8.02-7.77(m,6h,nap-h),7.57-7.47(m,2h,py-h),7.25-7.01(m,20h,ar-h),6.71(s,2h,ar-h),5.33(s,2h,ch),1.22(s,3h,c=n-ch3).
[0098]
实施例11
[0099]
吡啶亚胺化合物b5的合成
[0100]
按照实施例7中的合成方法,将实施例7中的n1替换为n5,产率65%。核磁氢谱如下:1h nmr(cdcl3,400mhz):δ8.53(s,1h,nap-h),8.25-8.16(d,1h,py-h),8.02-7.77(m,6h,nap-h),7.57-7.47(m,2h,py-h),7.25-7.01(m,20h,ar-h),6.71(s,2h,ar-h),5.33(s,2h,ch),1.23(s,3h,c=n-ch3).
[0101]
实施例12
[0102]
配体吡啶胺基化合物l1的合成
[0103]
氮气氛围下,用干燥的甲苯溶解吡啶亚胺化合物,在0℃下滴加四氢铝锂(1.1eq)溶液,回流过夜,分液、干燥,重结晶得到白色晶体5.65g,产率88%,为吡啶胺基配体l1。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.24-8.17(d,1h,ar-h),7.69-7.59(dd,2h,ar-h),7.44-7.39(d,1h,ar-h),7.27-7.18(m,7h,ar-h),7.11-6.93(m,18h,ar-h),6.87(s,2h,ar-h),6.46(dd,1h,ar-h),6.16(s,2h,chph2),4.59(d,1h,c-nh),4.38(m,1h,ch-n),1.85(s,3h,ar-ch3),1.55(d,3h,nc-ch3).
[0104]
实施例13
[0105]
配体吡啶胺基化合物l2的合成
[0106]
按照实施例12中的合成方法,将实施例12中的苯胺n1替换为n2,产率64%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.24-8.17(d,1h,ar-h),7.69-7.59(dd,2h,ar-h),7.44-7.39(d,1h,ar-h),7.27-7.18(m,7h,ar-h),7.11-6.93(m,18h,ar-h),6.87(s,2h,ar-h),6.46(dd,1h,ar-h),6.16(s,2h,chph2),4.59(d,1h,c-nh),3.48(s,1h,ar-och3),1.55(m,6h,nc(ch3)).
[0107]
实施例14
[0108]
配体吡啶胺基化合物l3的合成
[0109]
按照实施例12中的合成方法,将实施例12中的苯胺n1替换为n3,产率73%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.24-8.17(d,1h,ar-h),7.69-7.59(dd,2h,ar-h),7.44-7.39(d,1h,ar-h),7.27-7.18(m,7h,ar-h),7.11-6.93(m,18h,ar-h),6.87(s,2h,ar-h),6.46(dd,1h,ar-h),6.16(s,2h,chph2),4.59(d,1h,c-nh),3.48(s,1h,ar-och3),1.55(m,6h,nc(ch3)2).
[0110]
实施例15
[0111]
配体吡啶胺基化合物l4的合成
[0112]
按照实施例12中的合成方法,将实施例12中的苯胺n1替换为n4,产率77%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.24-8.17(d,1h,ar-h),7.69-7.59(dd,2h,ar-h),7.44-7.39(d,1h,ar-h),7.27-7.18(m,7h,ar-h),7.11-6.93(m,18h,ar-h),6.87(s,2h,ar-h),6.46(dd,1h,ar-h),6.33(s,2h,chph2),4.62(d,1h,c-nh),1.61(m,3h,nc(ch3)).
[0113]
实施例16
[0114]
配体吡啶胺基化合物l5的合成
[0115]
按照实施例12中的合成方法,将实施例12中的苯胺n1替换为n2,产率62%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.24-8.17(d,1h,ar-h),7.69-7.59(dd,2h,ar-h),7.44-7.39(d,1h,ar-h),7.27-7.18(m,7h,ar-h),7.11-6.93(m,18h,ar-h),6.87(s,2h,ar-h),6.46(dd,1h,ar-h),6.46(s,2h,chph2),4.67(d,1h,c-nh),1.65(m,3h,nc(ch3)).
[0116]
实施例17
[0117]
配体吡啶胺基化合物l6的合成
[0118]
按照实施例12中的合成方法,将实施例12中的苯胺n1替换为n6,产率86%。1h nmr(c6d6,400mhz):δ8.24-8.17(d,1h,ar-h),7.69-7.59(dd,2h,ar-h),7.44-7.39(d,1h,ar-h),7.27-7.18(m,7h,ar-h),7.11-6.93(m,18h,ar-h),6.87(s,2h,ar-h),6.46(dd,1h,ar-h),6.46(s,2h,chph2),4.67(d,1h,c-nh),1.49(m,6h,nc(ch3)2).
[0119]
二、铪配合物的制备
[0120]
实施例18
[0121]
配合物c1的合成
[0122]
称取l1(1.7mmol),用10ml甲苯溶解,0℃下滴加正丁基锂溶液(1.14ml,1.6m),反应3h。抽干甲苯,用正己烷洗涤,将上清液倒出,得到黄色的锂盐。用甲苯重新溶解锂盐,将hfcl4(0.61g,1.9mmol)转移进反应体系中,升温至90℃回流过夜。溶液温度随后降至室温,滴加memgbr溶液(2.13ml,3mol/l),室温搅拌3h。将溶剂抽干,用正己烷洗涤固体3次,过滤后收集正己烷滤液。将溶剂浓缩至约3ml,在-35℃下过夜结晶。将晶体过滤,用冷冻的正己烷洗涤、干燥。得到橙黄色晶体,产率46%。1h nmr(c6d6,400mhz):δ8.43(d,1h,ar-h),8.39(d,1h,ar-h),7.82(d,1h,ar-h),7.78(d,1h,ar-h),7.59(d,2h,ar-h),7.55(d,2h,ar-h),7.51(d,2h,ar-h),7.43-6.98(m,16h,ar-h),6.90-6.78(m,5h,ar-h),6.28(d,1h,ar-h),5.80(s,1h,hcph2),5.08(pseudo d,1h,nch),1.97(s,3h,ar-ch3),1.07(d,3h,cch3),0.82(s,3h,hf-ch3),0.37(s,3h,hf-ch3).
[0123]
实施例19
[0124]
配合物c2的合成
[0125]
按照实施例18中的合成方法,将实施例18中的配体l1替换为l2,产率35%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.44(d,1h,ar-h),8.39(d,1h,ar-h),7.82(d,1h,ar-h),7.78(d,1h,ar-h),7.59(d,2h,ar-h),7.55(d,2h,ar-h),7.51(d,2h,ar-h),7.43-6.98(m,16h,ar-h),6.90-6.78(m,5h,ar-h),6.28(d,1h,ar-h),5.80(s,1h,hcph2),5.08(pseudo d,2h,nch),1.11(d,3h,cch3),1.07(d,3h,och3),0.82(s,3h,hf-ch3),0.37(s,3h,hf-ch3).
[0126]
实施例20
[0127]
配合物c3的合成
[0128]
按照实施例18中的合成方法,将实施例18中的配体l1替换为l3,产率37%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.42(d,1h,ar-h),8.39(d,1h,ar-h),7.82(d,1h,ar-h),7.78(d,1h,ar-h),7.56(d,2h,ar-h),7.55(d,2h,ar-h),7.51(d,2h,ar-h),7.43-6.98(m,16h,ar-h),6.90-6.78(m,5h,ar-h),6.28(d,1h,ar-h),5.80(s,1h,hcph2),5.08(pseudo d,2h,nch),1.07(d,3h,cch3),0.82(s,3h,hf-ch3),0.38(s,3h,hf-ch3).
[0129]
实施例21
[0130]
配合物c4的合成
[0131]
按照实施例18中的合成方法,将实施例18中的配体l1替换为l4,产率27%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.42(d,1h,ar-h),8.39(d,1h,ar-h),7.82(d,1h,ar-h),7.78(d,1h,ar-h),7.56(d,2h,ar-h),7.55(d,2h,ar-h),7.51(d,2h,ar-h),7.43-6.98(m,16h,ar-h),6.90-6.78(m,5h,ar-h),6.28(d,1h,ar-h),5.80(s,1h,hcph2),5.08(pseudo d,2h,nch),1.07(d,3h,cch3),0.82(s,3h,hf-ch3),0.38(s,3h,hf-ch3).
[0132]
实施例22
[0133]
配合物c5的合成
[0134]
按照实施例18中的合成方法,将实施例18中的配体l1替换为l5,产率28%。核磁氢谱如下:1h nmr(c6d6,400mhz):δ8.45(d,1h,ar-h),8.39(d,1h,ar-h),7.84(d,1h,ar-h),7.78(d,1h,ar-h),7.56(d,2h,ar-h),7.55(d,2h,ar-h),7.51(d,2h,ar-h),7.43-6.96(m,16h,ar-h),6.90-6.78(m,5h,ar-h),6.28(d,1h,ar-h),5.80(s,1h,hcph2),5.08(pseudo d,2h,nch),1.07(d,3h,cch3),0.83(s,3h,hf-ch3),0.38(s,3h,hf-ch3).
[0135]
实施例23
[0136]
配合物c6的合成
[0137]
称取l6(1.5mmol),用10ml甲苯溶解,0℃下滴加正丁基锂溶液(1.04ml,1.6m),反应3h.抽干甲苯,用正己烷洗涤,将上清液倒出,得到黄色的锂盐。用甲苯重新溶解锂盐,将hfcl4(1.65mmol)用甲苯冲洗转移进体系中,升温至90℃回流过夜。降至室温,滴加memgbr溶液(2ml,3[m]),室温搅拌3h。将溶剂抽干,用正己烷洗涤固体3次,过滤后收集正己烷滤液.将溶剂浓缩至约3ml,在-35℃下过夜结晶。将晶体过滤,用冷冻的正己烷洗涤、干燥。得到橙黄色晶体,产率37%。1h nmr:(c6d6,400mhz):δ8.43(d,1h,ar-h),8.39(d,1h,ar-h),7.82(d,1h,ar-h),7.78(d,1h,ar-h),7.59(d,2h,ar-h),7.55(d,2h,ar-h),7.51(d,2h,ar-h),7.43-6.98(m,16h,ar-h),6.90-6.78(m,5h,ar-h),6.28(d,1h,ar-h),5.80(s,1h,hcph2),5.08(pseudo d,1h,nch),1.97(s,3h,ar-ch3),1.57(d,6h,c(ch3)2),0.82(s,3h,hf-ch3),0.37(s,3h,hf-ch3).
[0138]
三、催化烯烃聚合
[0139]
实施例24
[0140]
本实施例提供了一种吡啶胺基铪化合物c1催化乙烯均聚的方法,如下步骤:
[0141]
在无水无氧的条件下,向反应釜内加入100ml干燥的甲苯和500μmol三异丁基铝(hf:al=1:100),将5μmol的吡啶胺基铪化合物c1与6μmol的三苯碳鎓四(五氟苯)硼酸盐(hf:b=1:1.2)混合活化后加入反应釜内,通入10atm的乙烯,在80℃下进行乙烯均聚反应10min,用质量分数为5%的盐酸酸化乙醇中终止聚合,搅拌1h后过滤,用乙醇洗涤三次,在70℃下真空干燥12h,得到线性的聚乙烯聚合物。
[0142]
本实施例中吡啶胺基铪化合物c1的催化活性为1.21
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为635kg/mol,分子量分布指数为2.3,熔融温度为132.2℃。
[0143]
实施例25
[0144]
本实施例提供了一种吡啶胺基铪化合物c2催化乙烯均聚的方法,步骤与实施例24中相同。
[0145]
本实施例中吡啶胺基铪化合物c2的催化活性为1.43
×
106g pe/(mol hf
·
h),制
备得到的线性聚乙烯的重均分子量为448kg/mol,分子量分布指数为2.5,熔融温度为131.6℃。
[0146]
实施例26
[0147]
本实施例提供了一种吡啶胺基铪化合物c3催化乙烯均聚的方法,步骤与实施例24中相同。
[0148]
本实施例中吡啶胺基铪化合物c3的催化活性为0.89
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为278kg/mol,分子量分布指数为2.6,熔融温度为130.4℃。
[0149]
实施例27
[0150]
本实施例提供了一种吡啶胺基铪化合物c4催化乙烯均聚的方法,步骤与实施例24中相同。
[0151]
本实施例中吡啶胺基铪化合物c4的催化活性为0.56
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为147kg/mol,分子量分布指数为2.6,熔融温度为130.2℃。
[0152]
实施例28
[0153]
本实施例提供了一种吡啶胺基铪化合物c5催化乙烯均聚的方法,步骤与实施例24中相同。
[0154]
本实施例中吡啶胺基铪化合物c5的催化活性为0.15
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为89kg/mol,分子量分布指数为2.4,熔融温度为128.1℃。
[0155]
实施例29
[0156]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法,步骤与实施例24中相同。
[0157]
本实施例中吡啶胺基铪化合物c6的催化活性为1.65
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为752kg/mol,分子量分布指数为1.6,熔融温度为133.1℃。
[0158]
实施例30
[0159]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,聚合温度为100℃。
[0160]
本实施例中吡啶胺基铪化合物c6的催化活性为2.13
×
106g polymer/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为661kg/mol,分子量分布指数为1.7,熔融温度为131.7℃。
[0161]
实施例31
[0162]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,聚合温度为120℃。
[0163]
本实施例中吡啶胺基铪化合物c6的催化活性为1.03
×
106g polymer/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为314kg/mol,分子量分布指数为1.7,熔融温度为130.7℃。
[0164]
实施例32
[0165]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,助催化剂三苯碳鎓四(五氟苯)硼酸盐用量为5μmol(hf:b=1:1)。
[0166]
本实施例中吡啶胺基铪化合物c6的催化活性为1.33
×
106g polymer/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为648kg/mol,分子量分布指数为1.7,熔融温度为131.4℃。
[0167]
实施例33
[0168]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,助催化剂三苯碳鎓四(五氟苯)硼酸盐用量为7.5μmol(hf:b=1:1.5)。
[0169]
本实施例中吡啶胺基铪化合物c6的催化活性为1.61
×
106g polymer/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为659kg/mol,分子量分布指数为1.6,熔融温度为131.7℃。
[0170]
实施例34
[0171]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,助催化剂三苯碳鎓四(五氟苯)硼酸盐用量为25μmol(hf:b=1:5)。
[0172]
本实施例中吡啶胺基铪化合物c6的催化活性为1.63
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为542kg/mol,分子量分布指数为1.7,熔融温度为131.4℃。
[0173]
实施例35
[0174]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,助催化剂三(五氟苯)硼烷用量为7.5μmol(hf:b=1:1.5)。
[0175]
本实施例中吡啶胺基铪化合物c6的催化活性为1.32
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为551kg/mol,分子量分布指数为1.5,熔融温度为131.5℃。
[0176]
实施例36
[0177]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,助催化剂n,n-二甲基苯胺四(五氟苯基)硼酸盐用量为7.5μmol(hf:b=1:1.5)。
[0178]
本实施例中吡啶胺基铪化合物c6的催化活性为1.12
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为502kg/mol,分子量分布指数为1.7,熔融温度为131.4℃。
[0179]
实施例37
[0180]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,三异丁基铝的用量为1.25mmol(hf:al=1:250)。
[0181]
本实施例中吡啶胺基铪化合物c6的催化活性为1.24
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为473kg/mol,分子量分布指数为1.7,熔融温度为131.3℃。
[0182]
实施例38
[0183]
本实施例提供了一种吡啶胺基铪化合物c6催化乙烯均聚的方法。按照实施例24中的实验方法,三异丁基铝的用量为2.5mmol(hf:al=1:500)。
[0184]
本实施例中吡啶胺基铪化合物c6的催化活性为1.36
×
106g pe/(mol hf
·
h),制备得到的线性聚乙烯的重均分子量为459kg/mol,分子量分布指数为1.8,熔融温度为131.3℃。
[0185]
实施例39
[0186]
本实例提供了一种吡啶胺基铪催化剂c6催化丙烯均聚的方法。
[0187]
在无水无氧的条件下,向反应釜内加入100ml干燥的甲苯和500μmol三异丁基铝,将5μmol的吡啶胺基铪化合物c6与6μmol的三苯碳鎓四(五氟苯)硼酸盐混合活化5min后加入反应釜内,通入5atm的丙烯,在80℃下进行丙烯均聚反应30min,用质量分数为5%的盐酸酸化乙醇中终止聚合,搅拌10h后过滤,用乙醇洗涤三次,在70℃下真空干燥12h,得到聚丙烯聚合物。
[0188]
本实例中吡啶胺基铪催化剂c6的催化活性为0.96
×
106g pp/(mol hf
·
h),制备得到的聚丙烯重均分子量573kg/mol,分子量分布指数为3.3,等规度为95%,熔融温度为148.1℃。
[0189]
实施例40
[0190]
本实例提供了一种吡啶胺基铪催化剂c6催化1-己烯均聚的方法。
[0191]
在无水无氧的条件下,向充满氮气的反应瓶里加入6ml干燥的甲苯、4ml的1-己烯和1mmol的三异丁基铝,将10μmol的吡啶胺基铪化合物c6与12μmol的三苯碳鎓四(五氟苯)硼酸盐混合活化后加入反应瓶内,30℃下反应2h。用质量分数为5%的盐酸酸化乙醇中终止聚合,搅拌10h后过滤,用乙醇洗涤三次,在70℃下真空干燥12h,得到聚合物。
[0192]
本实例中吡啶胺基铪催化剂c6的催化活性为0.74
×
106g polymer/(mol hf
·
h),得到的聚合物重均分子量为18.4kg/mol,分子量分布指数为1.2,单体转化率为98%。
[0193]
实施例41
[0194]
本实例提供了一种吡啶胺基铪催化剂c6催化1-辛烯均聚的方法。
[0195]
在无水无氧的条件下,向充满氮气的反应瓶里加入6ml干燥的甲苯、4ml的1-辛烯和1mmol的三异丁基铝,将10μmol的吡啶胺基铪化合物c6与12μmol的三苯碳鎓四(五氟苯)硼酸盐混合活化后加入反应瓶内,30℃下反应2h。用质量分数为5%的盐酸酸化乙醇中终止聚合,搅拌10h后过滤,用乙醇洗涤三次,在70℃下真空干燥12h,得到聚合物。
[0196]
本实例中吡啶胺基铪催化剂c6的催化活性为0.67
×
106g polymer/(mol hf
·
h),得到的聚合物重均分子量为15.2kg/mol,分子量分布指数为1.3,单体转化率为94%。
[0197]
实施例42
[0198]
本实例提供了一种吡啶胺基铪催化剂c6催化乙烯和丙烯共聚的方法。
[0199]
在无水无氧的条件下,向反应釜内加入100ml干燥的甲苯、4g的丙烯和500μmol三异丁基铝,将5μmol的吡啶胺基铪化合物c6与6μmol的三苯碳鎓四(五氟苯)硼酸盐混合活化后加入反应釜内,通入10atm的乙烯,在80℃下进行共聚反应10min,用质量分数为5%的盐酸酸化乙醇中终止聚合,搅拌10h后过滤,用乙醇洗涤三次,在70℃下真空干燥12h,得到聚合物。
[0200]
本实例中吡啶胺基铪催化剂c6的催化活性为1.77
×
106g polymer/(mol hf
·
h),制备得到的共聚物重均分子量970.9kg/mol,分子量分布指数为3.1。
[0201]
实施例43
[0202]
本实例提供了一种吡啶胺基铪催化剂c6催化乙烯和1-己烯共聚的方法。
[0203]
在无水无氧的条件下,向反应釜内加入100ml干燥的甲苯、3.37g的1-己烯和500μmol三异丁基铝,将5μmol的吡啶胺基铪化合物c6与6μmol的三苯碳鎓四(五氟苯)硼酸盐混合活化后加入反应釜内,通入10atm的乙烯,在80℃下进行共聚反应10min,用质量分数为5%的盐酸酸化乙醇中终止聚合,搅拌10h后过滤,用乙醇洗涤三次,在70℃下真空干燥12h,得到聚合物。
[0204]
本实例中吡啶胺基铪催化剂c6的催化活性为2.03
×
106g polymer/(mol hf
·
h),制备得到的共聚物重均分子量847.6kg/mol,分子量分布指数为3.7,1-己烯插入率为18%,熔融温度为73.9℃和113.2℃。
[0205]
实施例44
[0206]
本实例提供了一种吡啶胺基铪催化剂c6催化乙烯和1-辛烯共聚的方法。
[0207]
在无水无氧的条件下,向反应釜内加入100ml干燥的甲苯、5.76g的1-辛烯和500μmol三异丁基铝,将5μmol的吡啶胺基铪化合物c6与6μmol的三苯碳鎓四(五氟苯)硼酸盐混合活化后加入反应釜内,通入10atm的乙烯,在80℃下进行共聚反应10min,用质量分数为5%的盐酸酸化乙醇中终止聚合,搅拌10h后过滤,用乙醇洗涤三次,在70℃下真空干燥12h,得到聚合物。
[0208]
本实例中吡啶胺基铪催化剂c6的催化活性为1.89
×
106g polymer/(mol hf
·
h),制备得到的共聚物重均分子量970.9kg/mol,分子量分布指数为3.4,1-辛烯插入率为15%,熔融温度为77.9℃和115.2℃。
[0209]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

