一种d
‑
a型共轭聚合物及其制备方法和应用
技术领域
1.本发明属于共轭聚合物材料技术领域,尤其涉及一种d
‑
a型共轭聚合物及其制备方法和应用。
背景技术:
2.近年来,随着石油、煤炭、天然气等不可再生资源的不断消耗,以及化石燃料燃烧带来的环境污染问题,让人们意识到开发新型的清洁可再生能源至关重要。结合太阳能取之不尽、用之不竭的特点以及氢能源清洁无污染、燃烧值大、可储存运输的优点,开发光催化水制氢的催化剂逐渐进入人们的视野。利用太阳光照射在催化剂上引起光子的跃迁,在一定的条件下分解水产生氢气,可实现太阳能到化学能的转变,且氢气也是一种清洁无污染的二次能源,有着广阔的应用前景。与生物制氢、矿物资源的重整等传统制氢手段相比,光催化分解水制氢更加符合绿色环保的要求,被誉为最为理想的产氢技术。
3.目前用于制氢的光催化剂主要是含金属的无机半导体,但其存在可见光活性低,金属资源有限等缺点。从可持续发展来看,纯有机半导体光催化剂在光催化分解水制氢领域中占据重要地位。d
‑
a型共轭聚合物作为一种新型的有机半导体光催化剂,由于其原料来源丰富、合成方法简单以及结构易于调控等独特的优势,引起了研究者们极大的关注。
4.但是目前的有机共轭聚合物作为光催化剂制氢的速率并不高,限制了其大规模应用。例如中国专利(cn111804338a)公开了一种三嗪基d
‑
a型含氮有机共轭多孔聚合物光催化材料及其制备与应用,主要采用的是三嗪基和吡唑小分子结合,从数据上看,其最高产氢速率(her)只有1000μmol
·
h
‑1·
g
‑1,无法与含金属的无机半导体媲美。
技术实现要素:
5.有鉴于此,本技术提供了一种d
‑
a型共轭聚合物及其制备方法和应用,提供了一种用于光催化分解水制氢时析氢速率高,化学稳定性和热稳定性好,且在可见光区展现了强的吸收的d
‑
a型共轭聚合物。
6.本发明第一方面提供了一种d
‑
a型共轭聚合物,所述d
‑
a型共轭聚合物的化学结构式如下所示:
[0007][0008]
其中,所述d
‑
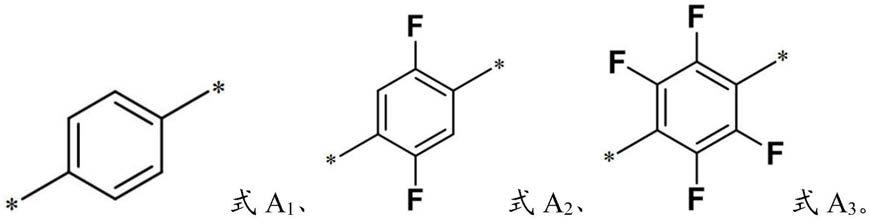
a型共轭聚合物的聚合度为50~250;所述选自式a1、式a2和式a3中的一种或多种,所述*为连接键位;
[0009][0010]
具体的,所述的*与所述d
‑
a型共轭聚合物的*连接。
[0011]
另一实施例中,所述d
‑
a型共轭聚合物的聚合度为180~250。
[0012]
具体的,所述d
‑
a型共轭聚合物的化学结构式如下所示:
[0013]
的一种或多种。
[0014]
本发明第二方面公开了所述的d
‑
a型共轭聚合物在光催化制氢中的应用;所述d
‑
a型共轭聚合物的化学结构式如下所示:
[0015][0016]
其中,所述d
‑
a型共轭聚合物的聚合度为50~250;所述选自式a1、式a2和式a3中的一种或多种,所述*为连接键位;
[0017][0018]
本发明第三方面公开了所述的d
‑
a型共轭聚合物在光催化制氢中的应用;所述d
‑
a型共轭聚合物的化学结构式如下所示:
[0019][0020]
其中,所述d
‑
a型共轭聚合物的聚合度为50~250;所述选自式4、式a5、式a6、式a7、式a8、式a9和式a
10
中的一种或多种,所述*为连接键位;
[0021][0022]
本发明第四方面提供了一种光催化水制氢的催化剂,所述催化剂的化学结构式如下所示:
[0023][0024]
其中,所述催化剂的聚合度为50~250;所述选自式a1、式a2和式a3中的一种或多种,所述*为连接键位;
[0025][0026]
另一实施例中,所述还选自式4、式a5、式a6、式a7、式a8、式a9和式a
10
中的一种或多种,所述*为连接键位;
[0027][0027][0028]
本发明第五方面公开了所述d
‑
a型共轭聚合物的制备方法,包括以下步骤:
[0029]
在惰性气氛下,将1,3,5
‑
三(噻吩
‑2‑
基)苯与单元、钯催化剂、无机碱和有机溶剂混合,进行加热反应,制得d
‑
a型共轭聚合物;
[0030]
所述选自式a1、式a2、式a3、式4、式a5、式a6、式a7、式a8、式a9和式a
10
中的一种或多种;
[0031][0031][0032]
具体的,本技术制备方法中,将反应原料与单元、钯催化剂、无机碱和有机溶剂混合,进行加热反应,制得d
‑
a型共轭聚合物;上述反应原料的结构式为所述单元的结构式为
[0033]
另一实施例中,所述钯催化剂为pd(pph3)4或/和pd(dppf)cl2。
[0034]
另一实施例中,所述加热反应的温度为100~160℃,所述加热反应的时间为12~60h。
[0035]
另一实施例中,所述1,3,5
‑
三(噻吩
‑2‑
基)苯与所述单元的摩尔比为2:3。
[0036]
另一实施例中,所述无机碱为碳酸钾或/和磷酸钾;所述所述有机溶剂选自氯苯、甲苯和n,n
‑
二甲基甲酰胺中的一种或多种。
[0037]
具体的,所述无机碱为碳酸钾;所述所述有机溶剂为dmf。
[0038]
另一实施例中,所述1,3,5
‑
三(噻吩
‑2‑
基)苯与所述钯催化剂的摩尔比为1:(0.01~0.015);所述1,3,5
‑
三(噻吩
‑2‑
基)苯与所述无机碱的摩尔比为1:(5~10)。
[0039]
本技术的d
‑
a型共轭聚合物具有多孔性,增加了将其用作光催化剂时催化反应的活性位点。本技术通过调整共轭聚合物的组成结构,改变了共轭聚合物的光谱响应范围和光学带隙,提高了共轭聚合物对太阳光的有效利用率,进而提高了本技术共轭聚合物用于光催化产氢的反应速率。此外,本技术以1,3,5
‑
三(噻吩
‑2‑
基)苯作为电子供体,与不同电子受体单元通过交叉偶联策略构筑得到d
‑
a型共轭聚合物,本技术共轭聚合物具有高化学稳定性和热稳定性,在可见光区展现了强的吸收,可高效利用太阳光,可在不添加任何助催化剂的情况下,实现高效光催化分解水制氢。
附图说明
[0040]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
[0041]
图1为本技术实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3的紫外
‑
可见吸收光谱图;
[0042]
图2为本技术实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3的热失重分析图;
[0043]
图3为本技术实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3的光催化水分解产氢效率图;
[0044]
图4为本技术实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3的红外光谱测试图。
具体实施方式
[0045]
本发明提供了一种d
‑
a型共轭聚合物及其制备方法和应用,用于解决现有技术中光催化聚合物存在的析氢速率低的技术缺陷。
[0046]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0047]
其中,以下实施例所用原料或试剂均为实收或自制。
[0048]
实施例1
[0049]
本技术实施例的d
‑
a型共轭聚合物的合成路线如下:
[0050][0051]
本技术实施例的d
‑
a型共轭聚合物的制备方法如下:
[0052]
(1)单体m1的合成:
[0053]
称量1,3,5
‑
三溴苯(10mmol,3.1480g),噻吩
‑2‑
基硼酸(35mmol,4.4786g)和pd(pph3)4(0.5mmol,0.5778g)加入250ml双颈烧瓶,抽换气三次除去反应瓶中的氧气。加入磷酸钾(2mmol
·
l
‑1,10ml)和无水thf(100ml)。在85℃下反应20h。用饱和nacl溶液洗涤并用二氯甲烷萃取,粗产物通过硅胶色谱柱纯化,并用四氢呋喃/甲醇重结晶,得白色固体1,3,5
‑
三(噻吩
‑2‑
基)苯;
[0054]
称量1,3,5
‑
三(噻吩
‑2‑
基)苯(1mmol,0.3245g)加入圆底烧瓶,加入thf(20ml);在0℃下,分批次加入nbs(4mmol,0.712g),避光条件下反应一晚。用硫代硫酸钠淬灭反应后用饱和nacl溶液洗涤并用二氯甲烷萃取,粗产物通过硅胶色谱柱纯化,并用四氢呋喃/甲醇重结晶,得微黄固体1,3,5
‑
三(5
‑
溴噻吩
‑2‑
基)苯;
[0055]
(2)共轭聚合物p1的合成:
[0056]
将单体m1(0.8mmol,0.4489g)和单体m2(1.2mmol,0.401g)加入48ml玻璃耐压瓶中;然后在充满氮气的手套箱中加入pd(pph3)4(0.025mmol,29mg),k2co3(2mol/l,6ml)以及dmf(20ml),拧紧四氟塞;在160℃条件下,反应48h。反应液冷却至室温后滴入甲醇溶液中,过滤得到粗产物,分别用甲醇、正己烷、二氯甲烷、四氢呋喃进行索氏抽提。剩余固体用甲醇冲洗,真空干燥24h,得到142.0mg黄绿色的d
‑
a型共轭聚合物cmp1。经测试,d
‑
a型共轭聚合物cmp1的聚合度为207。
[0057]
本技术实施例的光催化剂由d
‑
a型共轭聚合物cmp1组成。
[0058]
实施例2
[0059]
本技术实施例的d
‑
a型共轭聚合物的合成路线如下:
[0060][0061]
本技术实施例的d
‑
a型共轭聚合物的制备方法如下:
[0062]
(1)单体m3的合成:将dmf溶液中的1,4
‑
二溴
‑
2,5
‑
二氟苯(2g,7.36mmol)、蒎可二硼酯(3.98g,0.0157mol)和koac(3.15g,0.032mol)在反应器小瓶中氩气换气。净化混合物20分钟后,将pdcl2(dppf)(112mg,0.153mmol)添加到溶液混合物中。混合物在反应器中在160℃下加热12小时,随后冷却。产品用盐水、h2o和二氯甲烷提取,用mgso4干燥。将溶剂减压蒸发后,粗产物经柱层析纯化。用正己烷溶液重结晶得到纯白色结晶m3。
[0063]
(2)共轭聚合物p2的合成;
[0064]
将上述实施例制得的单体m1(0.8mmol,0.4489g)和单体m3(1.2mmol,0.4394g)加入48ml玻璃耐压瓶中;然后在充满氮气的手套箱中加入pd(pph3)4(0.025mmol,29mg),k2co3(2mol/l,6ml)以及dmf(20ml),拧紧四氟塞;在160℃条件下,反应48h。反应液冷却至室温后滴入甲醇溶液中,过滤得到粗产物,分别用甲醇、正己烷、二氯甲烷、四氢呋喃进行索氏抽提。剩余固体用甲醇冲洗,真空干燥24h,得到152.7mg黄色的d
‑
a型共轭聚合物cmp2。经测试,d
‑
a型共轭聚合物cmp2的聚合度为203。
[0065]
本技术实施例的光催化剂由d
‑
a型共轭聚合物cmp2组成。
[0066]
实施例3
[0067]
本技术实施例的d
‑
a型共轭聚合物的合成路线如下:
[0068][0069]
本实施例所述d
‑
a型共轭聚合物的制备方法如下:
[0070]
(1)单体m4的合成:
[0071]
在
‑
78℃氩气气氛下,将n
‑
buli(2.5m,正己烷,6.39ml,10.227mmol)滴加到1,4
‑
二溴四氟苯(1.5g,4.87mmol)的无水四氢呋喃(30ml)溶液中,搅拌1小时。之后在
‑
78℃下加入三甲基氯化锡(1.0m inthf,10.23ml,10.227mmol),在室温下搅拌反应混合物过夜。将溶液倒入水中,用乙醚提取有机层。除去溶剂,形成白色固体。石油醚再结晶得到白色固体晶体m4。
[0072]
(2)共轭聚合物p3的合成:
[0073]
准确称取上述实施例制得的单体m1(0.8mmol,0.4489g)和单体m4(1.2mmol,0.4626g),加入48ml厚壁耐压瓶中,在惰性气体环境下加入10ml甲苯,5wt%钯催化剂,密封。在120℃的条件下避光反应48h。反应液冷却至室温后,滴入不断搅拌的甲醇溶液中,过滤得到粗产物。粗产物依次用100ml甲醇、石油醚、二氯甲烷、氯苯溶剂进行索氏抽提各24h,剩余固体用甲醇冲洗,真空干燥24h,得到黄绿色的d
‑
a型共轭聚合物cmp3。经测试,d
‑
a型共轭聚合物cmp2的聚合度为203。
[0074]
本技术实施例的光催化剂由d
‑
a型共轭聚合物cmp3组成。
[0075]
实施例4
[0076]
本技术实施例的d
‑
a型共轭聚合物的合成路线如下:
[0077][0078]
本技术实施例的d
‑
a型共轭聚合物的制备方法如下:
[0079]
(1)单体m5的合成:
[0080]
称量3,7
‑
二溴二苯并[b,d]噻吩(1mmol,0.3425g),联硼酸频那醇酯(2.5mmol,0.635g),醋酸钾(4mmol,0.392g)和pd(dppf)cl2(0.05mmol,0.0366g)加入50ml两颈烧瓶,将反应物抽换气三次以除去氧气。加入干燥后的1,4
‑
二氧六环(15ml),再次将反应液抽换气,然后加热到80℃,反应12h。用饱和nacl溶液洗涤并用二氯甲烷萃取,粗产物通过硅胶色谱柱纯化,并用四氢呋喃/甲醇重结晶,得到白色晶体3,7
‑
双(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧杂硼烷
‑2‑
基)二苯并[b,d]噻吩(m5)。
[0081]
(2)共轭聚合物p4的合成:
[0082]
将上述实施例制得的单体m1(0.8mmol,0.4489g)和单体m5(1.2mmol,0.5234g)加入75ml玻璃耐压瓶中;然后在充满氮气的手套箱中加入pd(pph3)4(0.014mmol,16mg),k2co3(2mol/l,5ml)以及dmf(20ml),拧紧四氟塞;在120℃条件下,反应48h。反应液冷却至室温后滴入甲醇溶液中,过滤得到粗产物,分别用甲醇、正己烷、二氯甲烷、四氢呋喃进行索氏抽提。剩余固体用甲醇冲洗,真空干燥24h,得到558mg黄绿色的d
‑
a型共轭聚合物cmp4。经测试,d
‑
a型共轭聚合物cmp4的聚合度为196。
[0083]
本技术实施例的光催化剂由d
‑
a型共轭聚合物cmp4组成。
[0084]
实施例5
[0085]
本技术实施例的d
‑
a型共轭聚合物的合成路线如下:
[0086][0087]
本技术实施例的d
‑
a型共轭聚合物的制备方法如下:
[0088]
1)单体m6的合成:
[0089]
称量二苯并噻吩砜(10mmol,2.1625g)加入25ml圆底烧瓶中,加入70ml浓硫酸,搅拌均匀后,在0℃下,分批次加入nbs(21mmol,3.7380g);避光条件下反应24h后,将反应液倒入冰水中并搅拌,固体用水和甲醇洗至ph=7,通过ch3cl重结晶得3,7
‑
二溴二苯并[b,d]噻吩5,5
‑
二氧化物(白色固体);
[0090]
称量3,7
‑
二溴二苯并[b,d]噻吩5,5
‑
二氧化物(10mmol,3.7405g),联硼酸频那醇酯(30mmol,7.620g),醋酸钾(60mmol,5.880g)和pd(dppf)cl2(0.5mmol,0.3658g)加入250ml双颈烧瓶,抽换气三次以除去氧气;加入无水dmf(60ml),再次将反应液抽换气,然后加热到90℃,反应12h。用饱和nacl溶液洗涤并用二氯甲烷萃取,粗产物通过硅胶色谱柱纯化,并用四氢呋喃/甲醇重结晶,得到白色晶体3,7
‑
双(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧杂硼硼烷
‑2‑
基)二苯并[b,d]噻吩5,5
‑
二氧化物(m6);
[0091]
(2)共轭聚合物p5的合成:
[0092]
将单体m1(0.8mmol,0.4489g)和单体m6(1.2mmol,0.5618g)加入75ml玻璃耐压瓶中;然后在充满氮气的手套箱中加入pd(pph3)4(0.014mmol,16mg),k2co3(2mol/l,5ml)以及dmf(20ml),拧紧四氟塞;在120℃条件下,反应48h。反应液冷却至室温后滴入甲醇溶液中,过滤得到粗产物,分别用甲醇、正己烷、二氯甲烷、四氢呋喃进行索氏抽提。剩余固体用甲醇冲洗,真空干燥24h,得到574mg黄色的共轭聚合物p5。经测试,共轭聚合物p5的聚合度为195。
[0093]
本技术实施例的光催化剂由上述共轭聚合物p5组成。
[0094]
实施例6
[0095]
本技术实施例提供了实施例1~5的共轭聚合物的物理和化学性能测试试验,包括:
[0096]
1、实施例1~5的共轭聚合物的析氢速率试验:
[0097]
分别将5mg实施例1~5制得的光催化剂加入到50ml去离子水中,牺牲剂选用三乙胺,超声30min分散均匀,用氙灯模拟太阳光光照,光功率为318mw/cm2,进行光催化产氢测试。反应过程中利用循环冷却水使反应溶液的温度保持在5℃。产生的氢气在带有热导检测器的在线gc7900气相色谱仪上进行检测,测试结果见表1。
[0098]
表1实施例1~5制得的光催化剂的析氢速率(μmol
·
g
‑1·
h
‑1)
[0099] 实施例1实施例2实施例3实施例4实施例5析氢速率1878571386167261
[0100]
2、对实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3分别进行紫外
‑
可见吸收光谱、热失重、光催化水分解产氢效率和红外光谱测试,结果如图1~4所示。
[0101]
图1为实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3的紫外
‑
可见吸收光谱图。从图1中可知,本技术提供的d
‑
a型共轭聚合物在可见光范围内具有良好的吸收。
[0102]
图2为实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3的热失重分析图。从图2中可以看出,本技术提供的d
‑
a型共轭聚合物的热稳定性良好。
[0103]
图3为实施例1~3提供的d
‑
a型共轭聚合物cmp1、cmp2和cmp3的光催化水分解产氢速率图。从图3可知,本技术提供的d
‑
a型共轭聚合物具有较高的产氢速率,其中cmp3具有最高的产氢速率。
[0104]
图4说明了本技术实施例成功制得d
‑
a型共轭聚合物。
[0105]
综上所述,本技术实施例提供了以1,3,5
‑
三(噻吩
‑2‑
基)苯作为电子供体,与不同电子受体单元通过交叉偶联策略构筑制得d
‑
a型共轭聚合物,本技术的d
‑
a型共轭聚合物具有高化学稳定性和热稳定性,在可见光区展现了强的吸收,可高效利用太阳光,可在不添加任何助催化剂的情况下,实现高效光催化分解水制氢。
[0106]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

