刚性
‑
柔性的zif
‑
8/多巴胺协同增强型纸基摩擦材料及制备方法
技术领域
1.本发明属于摩擦材料技术领域,涉及一种刚性
‑
柔性的zif
‑
8/多巴胺协同增强型纸基摩擦材料及制备方法。
背景技术:
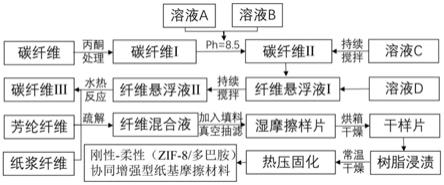
2.纸基摩擦材料是以碳纤维、芳纶纤维等增强纤维为主要原料,基于湿法成型与制浆造纸技术,经过真空抽滤和树脂热压固化制备而成。由于其具有摩擦系数稳定、磨损率低和工作寿命长等优点,而广泛应用于各类车辆和工程机械、机床、船舶等行业湿式离合器和制动器中。然而碳纤维表面能低、化学惰性强和与树脂界面结合性能差,导致在实际使用过程中,碳纤维增强纸基摩擦材料会出现碳纤维断裂和界面脱粘等现象,从而限制了此类材料在严苛的环境和特殊领域中的进一步应用。因此碳纤维与树脂浸润性差,两相间的界面脱粘,树脂剥离等问题已成为其瓶颈所在,严重制约了该类材料摩擦性能和使用寿命的提升。
3.文献1“专利公开号为的cn109338730a中国专利”公开了一种芳稠环分子组装改性碳纤维表面的方法及碳纤维界面增强树脂基复合材料的制备方法。该发明将碳纤维浸渍于芳稠环酰亚胺分子组装液中,得到表面改性的碳纤维。另外将改性碳纤维与树脂复合固化,最终得到芳稠环分子组装改性碳纤维界面增强树脂基复合材料。经过芳稠环分子改性碳纤维的化学活性得到一定程度的提高,同时也改善了树脂基复合材料的界面结合强度。然而此反应过程中使用有机溶剂体系包括n,n
‑
二甲基甲酰胺、甲苯、甲醇、乙腈和乙醚,会对环境造成严重的污染负担,不符合绿色可持续发展战略要求。
4.文献2“专利公开号为的cn110540662a中国专利”公开了一种聚多巴胺改性碳纤维/莫来石晶须增强树脂基摩擦材料的制备方法。该发明将碳纤维和莫来石晶须先进行脱胶处理,然后再通过多巴胺的自氧化聚合反应在其表面沉积纳米级聚多巴胺颗粒,将改性后的莫来石晶须和碳纤维与改性的酚醛树脂进行热压固化制备出树脂基摩擦材料。此方法有效地增强了树脂与碳纤维/莫来石晶须的界面结合强度,提高了摩擦材料的摩擦磨损性能和力学性能。由于莫来石晶须属于无机非金属矿物纤维,本身缺少活性基团,虽然本发明中采用多巴胺改性处理,只是在晶须表面形成了简单的物理包覆,因此在实际使用过程中,仍可能会出现界面脱粘和纤维断裂等问题。
5.文献3“专利公开号为的cn106868902a中国专利”公开了一种片状自组装二氧化锰改性碳纤维增强树脂基摩擦材料的制备方法。该发明将碳纤维进行浓硝酸氧化预处理,再使用高锰酸钾和浓硝酸基于氧化还原反应于碳纤维表面原位生长片状自组装二氧化锰。此方法有效增大纤维与树脂结合的比表面积,从而改善树脂基摩擦材料的摩擦磨损性能。然而通过强酸氧化处理纤维,引入活性官能团,一定程度上改善了碳纤维/基体的界面结合性能,但是会严重损伤纤维本身强度,弱化碳纤维增强效果。
6.上述所列文献中以及其它同领域专利文献大部分常采用强酸预处理碳纤维来改
善碳纤维的表面粗糙度和化学活性,但普遍存在对纤维本体产生严重损伤等问题。目前现有的技术中改善复合材料界面结合的方法单一,亟需迫切需要开发一种简单有效的方法来改善纸基摩擦材料的界面结合,同时保持纤维材料本体强度性能,从而综合提升纸基摩擦材料的摩擦磨损性能。目前,采用双组分zif
‑
8与多巴胺构筑刚性
‑
柔性界面结构协同增强纸基摩擦材料等方面并没有相关研究。
技术实现要素:
7.要解决的技术问题
8.为了避免现有技术的不足之处,本发明提出一种刚性
‑
柔性的zif
‑
8/多巴胺协同增强型纸基摩擦材料及制备方法,以克服现有技术存在的问题。首先采用多巴胺预处理碳纤维,达到活化碳纤维表面的目的,赋予惰性纤维表面部分活性反应基团。再利用原位生长技术,采用水热法将zif
‑
8纳米晶体生长在碳纤维表面,以获得zif
‑
8晶体修饰的碳纤维,再基于制浆造纸技术,通过真空抽滤方式得到湿摩擦样片,最后经过树脂浸渍和热压固化处理,从而制备出刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。由于多巴胺具有广谱粘性,可以在碳纤维基体上形成一层柔性纳米薄膜,同时多巴胺本身带有部分活性基团,可以活化碳纤维表面的同时为后续晶体生长提供有利的成核位点。进一步利用水热法于碳纤维表面原位生长zif
‑
8晶体,其晶体表面具有丰富的金属活性位点和大量的活性基团,双组分增强物质zif
‑
8/多巴胺构筑形成特殊的刚性
‑
柔性界面结构,不仅可以增强碳纤维与树脂基体之间的机械互锁作用,同时两者之间形成了有效化学耦合。最终,构筑刚性
‑
柔性的界面增强结构达到显著改善复合材料的层间结合效果,进一步提高纸基摩擦材料的摩擦磨损性能。
9.技术方案
10.一种刚性
‑
柔性的zif
‑
8/多巴胺协同增强型纸基摩擦材料,包括碳纤维、芳纶纤维和纸浆纤维;其特征在于:所述碳纤维表面生长有zif
‑
8纳米晶体的纳米金属骨架化合物。
11.一种制备所述刚性
‑
柔性的zif
‑
8/多巴胺协同增强型纸基摩擦材料的方法,其特征在于步骤如下:
12.步骤1:将碳纤维经过丙酮浸泡清洗,除去纤维表面的上浆剂和其它杂质,烘干得到表面干净的碳纤维i;
13.步骤2:室温条件下将三羟甲基氨基甲烷tris缓冲液溶解于去离子水中,进行磁力搅拌,获得溶液a,再将多巴胺溶解于去离子水中,进行超声分散,获得溶液b;
14.步骤3:采用酸调节溶液a的ph至8
‑
9,在磁力持续搅拌过程中,将溶液b倾倒入溶液a中充分混合;将步骤1中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘干得到碳纤维ii;
15.所述烘干温度为80
‑
120℃,烘干时间为12
‑
24h;
16.步骤4:室温条件下将六水合硝酸锌溶解于去离子水中,获得溶液c,再将2
‑
甲基咪唑分别溶解于去离子水中,获得溶液d;
17.步骤5:将步骤3中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
18.步骤6:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
19.步骤7:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴温度为60~85℃的水浴环境中,反应24~48h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥获得碳纤维iii;
20.所述干燥方式包括冷冻干燥:干燥温度为
‑
45~
‑
56℃,干燥时间为48~56h,干燥过程的真空度为15~25pa;烘箱干燥:干燥温度为70~105℃,干燥时间为12~24h;和常温干燥。
21.步骤8:将步骤7得到的zif
‑
8/多巴胺改性的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,加入填料,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥;
22.所述碳纤维iii︰芳纶纤维︰纸浆纤维︰填料的质量比为1.2~1.4︰1.0~1.2︰1.3~1.5︰2.5~3.2;上述质量百分比之和为100%;
23.所述烘箱干燥温度为50~75℃,干燥时间为1~4h
24.步骤9:将步骤8得到的物质于改性酚醛树脂中浸渍,完成后室温干燥,热压固化处理,从而获得刚性
‑
柔性的zif
‑
8/多巴胺协同增强型纸基摩擦材料;
25.所述热压固化处理的参数为:热压固化温度130~180℃,固化时间为3~15min,5~20pa;
26.所述改性酚醛树脂浸渍浓度为15~25%。
27.所述步骤1是将碳纤维置于丙酮溶液浸泡12~24h,烘干温度为80
‑
120℃,烘干时间为12
‑
24h。
28.所述步骤2溶液a中tris缓冲液中的质量浓度为1.0~1.5%。
29.所述步骤2的溶液b多巴胺的质量浓度为1.8~2.2%。
30.所述步骤3的酸调节液包括但不限于:盐酸、乙酸或硫酸。
31.所述步骤3的磁力搅拌转数为1200
‑
1500r。
32.所述步骤4的溶液c中六水合硝酸锌的质量浓度为8~13%。
33.所述步骤4的溶液d中2
‑
甲基咪唑的质量浓度为30~55%。
34.所述填料包括但不限于硫酸钡、石墨、三氧化二铝、铬铁矿、萤石粉、硅藻土、蒙脱土或滑石粉。
35.有益效果
36.本发明提出的一种刚性
‑
柔性的zif
‑
8/多巴胺协同增强型纸基摩擦材料及制备方法,将碳纤维经过丙酮浸泡清洗,除去纤维表面的上浆剂和其它杂质;将表面干净的碳纤维先后置于三羟甲基氨基甲烷(tris缓冲液)和多巴胺溶液中,在其表面形成一层高黏附的多巴胺柔性薄膜;随后通过绿色水热反应,于碳纤维表面原位生长致密且均匀的纳米zif
‑
8晶体刚性层,反应完成后将改性碳纤维充分洗涤,烘干。以改性后的碳纤维与树脂基体构筑刚性
‑
柔性的界面增强结构,制备zif
‑
8/多巴胺协同增强型纸基摩擦材料。本发明所制备的刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料动摩擦系数从0.1068提升到0.1356,增幅为26.97%,磨损率由3.55
×
10
‑8cm3·
j
‑1下降到2.45
×
10
‑8cm3·
j
‑1,降幅为30.99%。充分显示了zif
‑
8/多巴胺双组分增强和构筑的刚性
‑
柔性界面增强结构协同作用,应用于纸基
摩擦材料可以显著提升其摩擦磨损性能。
37.与现有技术相比,本发明具有以下有益的技术效果:
38.本发明通过zif
‑
8/多巴胺双组分协同增强,构筑刚性
‑
柔性的界面增强结构,制备高性能的纸基摩擦材料。基于机械啮合和化学耦合的协同机理,显著改善纤维与树脂基体之间的界面结合,最终达到提高复合材料的摩擦学性能目的。
39.(1)本发明采用纳米晶体zif
‑
8作为刚性单元,薄膜状多巴胺作为柔性单元设计一种特殊的刚性
‑
柔性界面增强结构,这种独特结构有利于纤维与基体良好结合,形成稳定的界面中间区域,从而防止纤维拔出和基体脱粘,另外,具有层次状的刚性
‑
柔性结构有利于传递外部应力,减少应力集中区,提高纸基摩擦材料抵抗应力损伤的能力。
40.(2)本发明采用多巴胺可以在碳纤维基体上形成一层纳米薄膜,同时多巴胺本身带有部分活性基团,从而显著活化碳纤维表面。相比于采用常规酸刻蚀预处理方式,多巴胺预处理纤维的优势在于:其包覆在纤维表面,赋予纤维部分活性基团的同时,不会对其表面和内部结构造成损伤,基本完整地保持碳纤维的本体强度,最终达到了活化碳纤维且保持高强度的双效预处理结果。另外包裹在碳纤维表面的多巴胺纳米薄膜,表面带有羧基和亚氨基等活性基团,可以在后续水热反应过程中为晶体zif
‑
8生长提供有利活性位点。
41.(3)本发明采用表面具有丰富的金属开放位点和大量的活性基团的zif
‑
8晶体增强,其作为桥梁物质,可以促使碳纤维和树脂基体之间达到强有力的机械互锁作用。相比于其它晶体,zif
‑
8原位增强纤维的优势在于:比表面积高达1224.18m2/g,有利于提高纤维与树脂之间的有效接触面积,同时其表面分布大量活性功能基团,进一步形成纤维与基体化学耦合作用。另外可以在摩擦材料表面形成致密的纳米耐磨层,改善材料的层间结合的同时,显著提高纸基摩擦材料的摩擦磨损性能。
42.本发明制备的刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料,相比于原始摩擦材料,动摩擦系数提高了26.97%;磨损率下降了30.99%。展现出了优异的摩擦磨损性能。
附图说明
43.图1是本发明的工艺流程示意图;
44.图2是空白实施例的原始碳纤维与本发明实施例1制备zif
‑
8/多巴胺协同修饰碳纤维的sem图(a
‑
空白实施例;b
‑
实施例1;a:
×
5000;b:
×
8000);
45.图3为在制动压力为0.5mpa,主轴转速为2000r/min的条件下测试的动摩擦系数;
46.图4是在制动压力为0.5mpa,主轴转速为2000r/min,制动次数为200次的条件下测试的磨损率。
具体实施方式
47.现结合实施例、附图对本发明作进一步描述:
48.参见图1,一种刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料及其制备方法,包括以下步骤:
49.步骤一:取15g碳纤维经丙酮浸泡12~24h,除去纤维表面的上浆剂和其它杂质,完成后使用去离子水冲洗3~5次,烘箱干燥,温度为80~120℃,时间为12~24h,烘干得到表
面干净的碳纤维i;
50.步骤二:室温条件下将1.0~1.5g三羟甲基氨基甲烷(tris缓冲液)溶解于0.97~1.45l去离子水中,进行磁力搅拌,获得溶液a,再将1.8~2.2g多巴胺溶解于0.03~0.05l去离子水中,进行超声分散,获得溶液b;
51.步骤三:采用盐酸调节溶液a的ph至8~9,在磁力持续搅拌过程中,磁力搅拌转数为1200~1500r,将溶液b倾倒入溶液a中,使其充分混合。将步骤一中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘箱干燥,温度为80~120℃,时间为12~24h,得到碳纤维ii备用;
52.步骤四:室温条件下将1.04~1.34g六水合硝酸锌溶解于9~12g去离子水中,获得溶液c,其中六水合硝酸锌的质量浓度为8~13%;再将17.14~36.67g 2
‑
甲基咪唑分别溶解于30~40g去离子水中,获得溶液d,其中2
‑
甲基咪唑的质量浓度为30~55%;
53.步骤五:将步骤三中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
54.步骤六:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
55.步骤七:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴环境中反应,水浴温度为60~85℃,反应时间为24~48h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥备用,可采用冷冻干燥(干燥温度为
‑
45~
‑
56℃,干燥时间为48~56h,干燥过程的真空度为15~25pa)、烘箱干燥(干燥温度为70~105℃,干燥时间为12~24h)和常温干燥。获得碳纤维iii;
56.步骤八:将步骤七得到的负载zif
‑
8晶体的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维iii、芳纶纤维、纸浆纤维和填料的质量比为1.2~1.4:1.0~1.2:1.3~1.5:2.5~3.2,上述质量百分比之和为100%。加入填料包括但不限于硫酸钡、石墨、三氧化二铝、铬铁矿、萤石粉、硅藻土、蒙脱土和滑石粉,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为50~75℃,时间为1~4h,获得干样片备用;
57.步骤九:将步骤八得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为15~25%,完成后室温干燥,热压固化处理,热压温度130~180℃,固化时间为3~15min,压力为5~20pa。从而获得刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。
58.下面结合实施例对本发明做进一步详细描述:
59.空白实施例
60.步骤一:将碳纤维、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维、芳纶纤维、纸浆纤维和填料的质量比为1.2:1.0:1.3:3.0,加入填料包括硫酸钡、石墨、三氧化二铝和滑石粉,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为75℃,时间为4h,获得干样片备用;
61.步骤二:将步骤一得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为20%,完成后室温干燥,热压固化处理,热压温度130℃,固化时间为3min,压力为15pa。从而获得空白纸基摩擦材料。
62.实施例1
63.步骤一:取15g碳纤维经丙酮浸泡12h,除去纤维表面的上浆剂和其它杂质,完成后使用去离子水冲洗3次,烘箱干燥,温度为80℃,时间为12h,烘干得到表面干净的碳纤维i;
64.步骤二:室温条件下将1.0g三羟甲基氨基甲烷(tris缓冲液)溶解于0.97l去离子水中,进行磁力搅拌,获得溶液a,再将1.8g多巴胺溶解于0.03l去离子水中,进行超声分散,获得溶液b;
65.步骤三:采用盐酸调节溶液a的ph至8,在磁力持续搅拌过程中,磁力搅拌转数为1200r,将溶液b倾倒入溶液a中,使其充分混合。将步骤一中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘箱干燥,温度为80℃,时间为12h,得到碳纤维ii备用;
66.步骤四:室温条件下将1.04g六水合硝酸锌溶解于9g去离子水中,获得溶液c,其中六水合硝酸锌的质量浓度为8%;再将17.14g 2
‑
甲基咪唑分别溶解于30g去离子水中,获得溶液d,其中2
‑
甲基咪唑的质量浓度为30%;
67.步骤五:将步骤三中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
68.步骤六:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
69.步骤七:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴环境中反应,水浴温度为60℃,反应时间为24h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥备用,采用冷冻干燥(干燥温度为
‑
56℃,干燥时间为48h,干燥过程的真空度为15pa)。获得碳纤维iii;
70.步骤八:将步骤七得到的负载zif
‑
8晶体的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维iii、芳纶纤维、纸浆纤维和填料的质量比为1.2:1.0:1.3:3.0,上述质量百分比之和为100%。加入填料包括硫酸钡、石墨、三氧化二铝和滑石粉,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为75℃,时间为4h,获得干样片备用;
71.步骤九:将步骤八得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为20%,完成后室温干燥,热压固化处理,热压温度130℃,固化时间为3min,压力为15pa。从而获得刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。
72.实施例2
73.步骤一:取15g碳纤维经丙酮浸泡16h,除去纤维表面的上浆剂和其它杂质,完成后使用去离子水冲洗3次,烘箱干燥,温度为85℃,时间为18h,烘干得到表面干净的碳纤维i;
74.步骤二:室温条件下将1.2g三羟甲基氨基甲烷(tris缓冲液)溶解于0.97l去离子水中,进行磁力搅拌,获得溶液a,再将1.9g多巴胺溶解于0.03l去离子水中,进行超声分散,获得溶液b;
75.步骤三:采用盐酸调节溶液a的ph至8.5,在磁力持续搅拌过程中,磁力搅拌转数为1300r,将溶液b倾倒入溶液a中,使其充分混合。将步骤一中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘箱干燥,温度为90℃,时间为16h,得到碳纤维ii备用;
76.步骤四:室温条件下将1.14g六水合硝酸锌溶解于10g去离子水中,获得溶液c,其中六水合硝酸锌的质量浓度为11.4%;再将18g 2
‑
甲基咪唑分别溶解于35g去离子水中,获
得溶液d,其中2
‑
甲基咪唑的质量浓度为51.4%;
77.步骤五:将步骤三中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
78.步骤六:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
79.步骤七:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴环境中反应,水浴温度为70℃,反应时间为30h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥备用,采用冷冻干燥(干燥温度为
‑
45℃,干燥时间为50h,干燥过程的真空度为20pa)。获得碳纤维iii;
80.步骤八:将步骤七得到的负载zif
‑
8晶体的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维iii、芳纶纤维、纸浆纤维和填料的质量比为1.2:1.2:1.3:2.8,上述质量百分比之和为100%。加入填料包括硫酸钡、石墨、三氧化二铝和滑石粉,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为75℃,时间为2h,获得干样片备用;
81.步骤九:将步骤八得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为15%,完成后室温干燥,热压固化处理,热压温度140℃,固化时间为5min,压力为15pa。从而获得刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。
82.实施例3
83.步骤一:取15g碳纤维经丙酮浸泡24h,除去纤维表面的上浆剂和其它杂质,完成后使用去离子水冲洗5次,烘箱干燥,温度为120℃,时间为24h,烘干得到表面干净的碳纤维i;
84.步骤二:室温条件下将1.2g三羟甲基氨基甲烷(tris缓冲液)溶解于0.97l去离子水中,进行磁力搅拌,获得溶液a,再将2.0g多巴胺溶解于0.03l去离子水中,进行超声分散,获得溶液b;
85.步骤三:采用盐酸调节溶液a的ph至8.5,在磁力持续搅拌过程中,磁力搅拌转数为1200r,将溶液b倾倒入溶液a中,使其充分混合。将步骤一中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘箱干燥,温度为105℃,时间为12h,得到碳纤维ii备用;
86.步骤四:室温条件下将1.34g六水合硝酸锌溶解于12g去离子水中,获得溶液c,其中六水合硝酸锌的质量浓度为11.17%;再将20g 2
‑
甲基咪唑分别溶解于40g去离子水中,获得溶液d,其中2
‑
甲基咪唑的质量浓度为50%;
87.步骤五:将步骤三中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
88.步骤六:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
89.步骤七:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴环境中反应,水浴温度为85℃,反应时间为30h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥备用,可采用烘箱干燥(干燥温度为105℃,干燥时间为24h)。获得碳纤维iii;
90.步骤八:将步骤七得到的负载zif
‑
8晶体的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维iii、芳纶纤维、纸浆纤维和填料的质量比为1.4:1.0:1.3:3.1,上述质量百分比之和为100%。加入填料包括铬铁矿、萤石粉、硅藻土、蒙脱土和滑石粉,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为75℃,时间为4h,获得干样片备用;
91.步骤九:将步骤八得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为18%,完成后室温干燥,热压固化处理,热压温度180℃,固化时间为5min,压力为10pa。从而获得刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。
92.实施例4
93.步骤一:取15g碳纤维经丙酮浸泡20h,除去纤维表面的上浆剂和其它杂质,完成后使用去离子水冲洗3次,烘箱干燥,温度为120℃,时间为24h,烘干得到表面干净的碳纤维i;
94.步骤二:室温条件下将1.5g三羟甲基氨基甲烷(tris缓冲液)溶解于0.97l去离子水中,进行磁力搅拌,获得溶液a,再将2.2g多巴胺溶解于0.03l去离子水中,进行超声分散,获得溶液b;
95.步骤三:采用盐酸调节溶液a的ph至9,在磁力持续搅拌过程中,磁力搅拌转数为1200r,将溶液b倾倒入溶液a中,使其充分混合。将步骤一中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘箱干燥,温度为120℃,时间为24h,得到碳纤维ii备用;
96.步骤四:室温条件下将1.04g六水合硝酸锌溶解于12g去离子水中,获得溶液c,其中六水合硝酸锌的质量浓度为8.7%;再将17.14g 2
‑
甲基咪唑分别溶解于40g去离子水中,获得溶液d,其中2
‑
甲基咪唑的质量浓度为42.8%;
97.步骤五:将步骤三中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
98.步骤六:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
99.步骤七:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴环境中反应,水浴温度为85℃,反应时间为48h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥备用,可采用烘箱干燥(干燥温度为70℃,干燥时间为12h)。获得碳纤维iii;
100.步骤八:将步骤七得到的负载zif
‑
8晶体的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维iii、芳纶纤维、纸浆纤维和填料的质量比为1.4:1.2:1.5:3.0,上述质量百分比之和为100%。加入填料包括铬铁矿、萤石粉、硅藻土和滑石粉,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为75℃,时间为4h,获得干样片备用;
101.步骤九:将步骤八得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为25%,完成后室温干燥,热压固化处理,热压温度180℃,固化时间为15min,压力为15pa。从而获得刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。
102.实施例5
103.步骤一:取15g碳纤维经丙酮浸泡12h,除去纤维表面的上浆剂和其它杂质,完成后
使用去离子水冲洗5次,烘箱干燥,温度为120℃,时间为24h,烘干得到表面干净的碳纤维i;
104.步骤二:室温条件下将1.5g三羟甲基氨基甲烷(tris缓冲液)溶解于1.45l去离子水中,进行磁力搅拌,获得溶液a,再将2.2g多巴胺溶解于0.05l去离子水中,进行超声分散,获得溶液b;
105.步骤三:采用盐酸调节溶液a的ph至9,在磁力持续搅拌过程中,磁力搅拌转数为1500r,将溶液b倾倒入溶液a中,使其充分混合。将步骤一中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘箱干燥,温度为120℃,时间为24h,得到碳纤维ii备用;
106.步骤四:室温条件下将1.04g六水合硝酸锌溶解于11g去离子水中,获得溶液c,其中六水合硝酸锌的质量浓度为9.5%;再将17.14g 2
‑
甲基咪唑分别溶解于40g去离子水中,获得溶液d,其中2
‑
甲基咪唑的质量浓度为42.8%;
107.步骤五:将步骤三中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
108.步骤六:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
109.步骤七:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴环境中反应,水浴温度为85℃,反应时间为48h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥备用,可采用冷冻干燥(干燥温度为
‑
50℃,干燥时间为50h,干燥过程的真空度为22pa)。获得碳纤维iii;
110.步骤八:将步骤七得到的负载zif
‑
8晶体的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维iii、芳纶纤维、纸浆纤维和填料的质量比为1.2:1.0:1.3:3.2,上述质量百分比之和为100%。加入填料包括铬铁矿、萤石粉、硅藻土和滑石粉,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为75℃,时间为4h,获得干样片备用;
111.步骤九:将步骤八得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为20%,完成后室温干燥,热压固化处理,热压温度160℃,固化时间为3min,压力为15pa。从而获得刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。
112.实施例6
113.步骤一:取15g碳纤维经丙酮浸泡24h,除去纤维表面的上浆剂和其它杂质,完成后使用去离子水冲洗5次,烘箱干燥,温度为120℃,时间为24h,烘干得到表面干净的碳纤维i;
114.步骤二:室温条件下将1.5g三羟甲基氨基甲烷(tris缓冲液)溶解于1.45l去离子水中,进行磁力搅拌,获得溶液a,再将2.2g多巴胺溶解于0.05l去离子水中,进行超声分散,获得溶液b;
115.步骤三:采用盐酸调节溶液a的ph至8.5,在磁力持续搅拌过程中,磁力搅拌转数为1500r,将溶液b倾倒入溶液a中,使其充分混合。将步骤一中得到碳纤维i置于上述混合溶液中,常温搅拌,反应完成后烘箱干燥,温度为120℃,时间为24h,得到碳纤维ii备用;
116.步骤四:室温条件下将1.04g六水合硝酸锌溶解于12g去离子水中,获得溶液c,其中六水合硝酸锌的质量浓度为8.7%;再将17.14g 2
‑
甲基咪唑分别溶解于40g去离子水中,获得溶液d,其中2
‑
甲基咪唑的质量浓度为42.8%;
117.步骤五:将步骤三中碳纤维ii置于溶液c中,不断搅拌,充分混合,使zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键,从而得到纤维悬浮液i;
118.步骤六:将纤维悬浮液i搅拌均匀后,立即将溶液d倾倒入纤维悬浮液i中,继续搅拌,从而获得纤维悬浮液ii;
119.步骤七:将悬浮液ii倾倒于聚四氟乙烯反应釜中,在水浴环境中反应,水浴温度为75℃,反应时间为24h,反应结束后,通过真空抽滤法分离出负载zif
‑
8晶体的纤维,并利用去离子水对负载zif
‑
8晶体的纤维清洗若干次,干燥备用,采用冷冻干燥(干燥温度为
‑
56℃,干燥时间为56h,干燥过程的真空度为25pa)。获得碳纤维iii;
120.步骤八:将步骤七得到的负载zif
‑
8晶体的碳纤维iii、芳纶纤维和纸浆纤维经过疏解配置成纤维混合液,碳纤维iii、芳纶纤维、纸浆纤维和填料的质量比为1.4:1.2:1.5:2.5,上述质量百分比之和为100%。加入填料包括硫酸钡、石墨、三氧化二铝、萤石粉和蒙脱土,通过真空抽滤的原理和制浆造纸的方式制得湿样片,置于烘箱干燥,温度为75℃,时间为3h,获得干样片备用;
121.步骤九:将步骤八得到的干样品于改性酚醛树脂中浸渍,浸渍浓度为20%,完成后室温干燥,热压固化处理,热压温度180℃,固化时间为3min,压力为20pa。从而获得刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料。
122.图1为本发明的工艺流程图。图2则是空白实施例中的原始碳纤维与实施例1中zif
‑
8/多巴胺协同修饰碳纤维的微观形貌对比。从图2
‑
a可以看出,原始碳纤维表面光滑,且表面缺乏活性基团,因此直接采用惰性较强的原始碳纤维制备纸基摩擦材料,导致碳纤维无法与基体之间形成有效结合,在高温高压的实际使用环境中,易发生纤维脱落和基体脱粘等问题,造成材料的摩擦性能下降。而由图2
‑
b可以明显看出,zif
‑
8晶体已成功的包裹在碳纤维的表面,由于经过碳纤维经过多巴胺预处理,在其表面形成一层具有广谱黏性的纳米薄膜,且薄膜表面存在的活性基团为后续纳米zif
‑
8晶体形成提供了有利的生长位点,zn
2
通过化学键和作用与碳纤维ii表面的多巴胺活性层的游离羟基反应,在碳纤维形成zn
‑
o键。如图所示在碳纤维表面形成均匀且致密纳米晶体zif
‑
8刚性耐磨层,可以进一步促使碳纤维与树脂基体形成强有力的机械互锁作用,同时产生有效的化学耦合效果,改善材料的层间结合的同时,显著提高纸基摩擦材料的摩擦磨损性能。
123.图3为在制动压力为0.5mpa,主轴转速为2000r/min的条件下,纸基摩擦材料测试的动摩擦系数。由图可以明显看出:相比于空白样品,刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料的动摩擦系数呈现上升趋势,其从0.1068提升到0.1356,增幅为26.97%。当碳纤维经过zif
‑
8和多巴胺修饰后,纸基摩擦材料的动摩擦系数大幅提升,这主要归因于原位生长致密的纳米zif
‑
8晶体层,形成纳米尺度凸起结构,显著改善材料表面的粗糙度,而粗糙度的提高导致摩擦副与试样之间形成更紧密的接触和更强的机械互锁,从而需要更大的摩擦力矩来克服上述机械啮合,导致动摩擦系数升高。
124.图4为在制动压力为0.5mpa,主轴转速为2000r/min,制动次数为200次的条件下测试的磨损率。由图可以明显看出:相比于空白样品,刚性
‑
柔性(zif
‑
8/多巴胺)协同增强型纸基摩擦材料的磨损率呈现下降趋势,其从3.55
×
10
‑8cm3·
j
‑1下降到2.45
×
10
‑8cm3·
j
‑1,降幅为30.99%。这主要是因为独特的刚性
‑
柔性层次结构有利于增强界面结合,形成稳定的
中间区域,防止纤维拔出和基体脱粘,同时这种刚柔结构有利于传递外部应力,减少应力集中区,提高复合材料抵抗应力损伤的能力。基于机械互锁与化学耦合的作用效果,改善材料内部层间结合,明显提高纸基摩擦材料的耐磨性能。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。