一种3
‑
氧代环丁烷基羧酸的制备方法
技术领域
1.本发明涉及一种3
‑
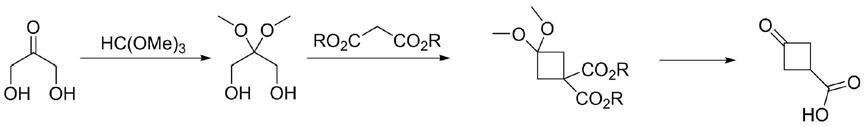
氧代环丁烷基羧酸的制备方法,属于医药中间体合成技术领域。
背景技术:
[0002]3‑
氧代环丁烷基羧酸,英文名:3
‑
oxocyclo butane carboxylic acid,cas:23761
‑
23
‑
1,是合成许多抗病毒药物和重要有机中间体的原料,在化学、化工、医药等领域有非常广泛的应用。
[0003]3‑
氧代环丁烷基羧酸是药物合成的关键原料,药物中间体对新药研发的发展十分重要,药物中间体是一类高科技密度,高附加值,用途专一的化工产品。3
‑
氧代环丁烷基羧酸可用于铂系药物中,其中环丁烷具有特殊的生物活性,羧基可与不同的多肽,氨基酸、氮杂环核苷类缩合得到不用的药物中间体,其药物用于激酶抑制剂、拮抗剂等在自身免疫疾病及抗肿瘤药物。
[0004]
关键步骤3,3
‑
二甲基环丁基
‑
1,1
‑
二羧酸二异丙酯的合成。其中us2009/233903,2009,a1收率56.32%,采用1,3
‑
二溴
‑
2,2
‑
二甲氧基丙烷与丙二酸二异丙酯与氢化钠70℃反应,并在dmf溶剂中高温140℃回流关环48小时。该方法中采用dmf与nah体系存在爆炸风险,同时高温长时间反应能耗较大。
[0005]
因此有必要开发合适的合成方法,避免采用高温,减少反应时间,收率高、反应条件温合、安全稳定的反应路线,并且适合工业放大生产的制备方法。
技术实现要素:
[0006]
为了满足工业化需求,本发明公开了3
‑
氧代环丁烷基羧酸的制备方法,以1,3
‑
二羟基丙酮为原料,与原甲酸三甲酯缩合对酮进行保护得到2,2
‑
二甲氧基
‑
1,3
‑
丙二醇,接着与丙二酸二酯在三苯基磷/偶氮二羧酸酯体系中反应得到3,3
‑
二甲基环丁基
‑
1,1
‑
二羧酸二异丙酯。随后在酸性条件下水解、脱羧及脱保护得到3
‑
氧代环丁烷基羧酸。
[0007]
本发明所述一种3
‑
氧代环丁烷基羧酸的制备方法,包括如下步骤:
[0008][0009]
第一步:将1,3
‑
二羟基丙酮、原甲酸三甲酯加入到甲醇溶液中,再加入催化剂反应,减压蒸馏得到2,2
‑
二甲氧基
‑
1,3
‑
丙二醇;
[0010]
第二步:氮气保护下,将三苯基磷、丙二酸二酯和碱溶于有机溶剂中,滴加2,2
‑
二甲氧基
‑
1,3
‑
丙二醇,接着加入偶氮二羧酸酯后升温反应,经后处理得到3,3
‑
二甲氧基环丁烷
‑
1,1
‑
二羧酸二酯;
[0011]
第三步:将3,3
‑
二甲氧基环丁烷
‑
1,1
‑
二羧酸二酯与相转移催化剂混合在盐酸中加热回流,随后重结晶得到3
‑
氧代环丁烷基羧酸。
[0012]
进一步地,在上述技术方案中,第一步中所述催化剂选自对甲苯磺酸,用量为原料重量的1
‑
3%。
[0013]
进一步地,在上述技术方案中,1,3
‑
二羟基丙酮与原甲酸三甲酯摩尔比为1:1.5
‑
2.0。
[0014]
进一步地,在上述技术方案中,第二步中,所述有机溶剂选自无水四氢呋喃或1,4
‑
二氧六环,所述偶氮二羧酸酯选自偶氮二羧酸二异丙酯或偶氮二甲酸二乙酯;丙二酸二酯选自丙二酸二异丙酯或丙二酸二乙酯。
[0015]
进一步地,在上述技术方案中,第二步中,所述2,2
‑
二甲氧基
‑
1,3
‑
丙二醇、丙二酸二酯、三苯基磷与偶氮二羧酸酯摩尔比为1:1.01
‑
1.05:2.1
‑
2.3:2.1
‑
2.3。
[0016]
进一步地,在上述技术方案中,第二步中,所述碱选自三乙胺、二异丙基乙基胺、t
‑
buok或dbu。
[0017]
进一步地,在上述技术方案中,第二步中,所述反应温度为
‑
5~50℃。
[0018]
进一步地,在上述技术方案中,第二步中,所述后处理将粗产品溶于正庚烷或石油醚中,通过预先铺装好少量硅胶的砂芯漏斗里过滤,可将副产物三苯基氧磷除掉。第二步中间体颜色及纯度由很大提升。
[0019]
进一步地,在上述技术方案中,第三步中,所述相转移催化剂选自甲基三辛基氯化铵,反应温度选自70
‑
80℃。
[0020]
进一步地,在上述技术方案中,第三步中,反应过程保持ph=1.5
‑
2.0;重结晶选自甲苯和正庚烷混合比例2:1对粗品进行提纯。
[0021]
发明有益效果
[0022]
本发明原料易得,操作简单,第二步选择更好的溶剂及快速过少量硅胶从而提升产品质量及颜色,有效去除了副产物三苯基氧磷及极性大的色素。第三步通过加入甲基三辛基氯化铵相转移催化剂,大大缩短了反应时间,避免使用如氢化钠这种易自燃易爆强亲核性试剂。使得本工艺更具备工业化放大前景。
具体实施例
[0023]
下面通过具体实例对本发明进行进一步说明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修改同样落入本发明权利要求所限定的范围。
[0024]
实施例1
[0025][0026]
氮气保护下,反应瓶内1,3
‑
二羟基丙酮45g(0.5mol)、无水的对甲苯磺酸4.5g和甲醇500ml。升温35℃,滴加原甲酸三甲酯79.6g(0.75mol),滴加结束后回流搅拌4小时,tlc检测磷钼酸显色无原料剩余,加入阻聚剂,改为常压蒸馏装置,蒸出大部分甲醇。最后65
‑
80℃减压蒸馏出前馏份,88~97℃下减压蒸馏(
‑
0.095~
‑
0.098mpa)得到无色油状物2,2
‑
二甲氧基
‑
1,3
‑
丙二醇59g,gc 98.8%,收率86.7%。1h nmr(400mhz,cdcl3)δ:4.12(s,2h),
3.78
‑
3.75(m,4h),3.29(s,6h).
[0027]
实施例2
[0028][0029]
在氮气保护下,向反应瓶内投入173.2g(0.66mol)三苯基磷、40.9g(0.3mol)2,2
‑
二甲氧基
‑
1,3
‑
丙二醇、59.3g(0.315mol)丙二酸二异丙酯和800ml无水四氢呋喃,降温至0℃,滴加三乙胺45.5g(0.45mol),搅拌30分钟,接着滴加diad 133.4g(0.66mol),随后升温至50℃反应6小时。加入氯化铵水溶液淬灭,分层,水相用正庚烷萃取,合并有机相,减压浓缩,正庚烷替换,有大量固体析出,通过预先铺装好少量硅胶的砂芯漏斗里过滤,用正庚烷淋洗多次,滤液减压浓缩至不流液,得到粗品3,3
‑
二甲氧基环丁烷
‑
1,1
‑
二羧酸二异丙酯71.9g,收率83.1%,gc 96.9%。1hnmr(400mhz,cdcl3):5.06
‑
5.04(m,2h),3.20(s,6h),2.70(m,4h),1.24
‑
1.20(m,12h).
[0030]
实施例3
[0031][0032]
在氮气保护下,向反应瓶内投入173.2g(0.66mol)三苯基磷、40.9g(0.3mol)2,2
‑
二甲氧基
‑
1,3
‑
丙二醇、50.5g(0.315mol)丙二酸二乙酯和800ml1,4
‑
二氧六环,降温至0℃,分批加入叔丁醇钾40.4g(0.45mol),加毕,室温搅拌1.5小时,接着滴加115g(0.66mol)dead。随后升温至50℃反应4小时。加入到冰水中,采用醋酸调节ph=6
‑
7。分层,水相用正庚烷萃取,合并有机相,减压浓缩,正庚烷替换,有大量固体析出,通过预先铺装好少量硅胶的砂芯漏斗里过滤,用正庚烷淋洗多次,滤液减压浓缩至不流液,得到粗品3,3
‑
二甲氧基环丁烷
‑
1,1
‑
二羧酸二乙酯57.7g,收率73.9%,gc 97.3%。1hnmr(400mhz,cdcl3):4.15
‑
4.12(m,4h),3.30(s,6h),2.82(m,4h),1.20
‑
1.18(m,6h).
[0033]
实施例4
[0034][0035]
在氮气保护下,向反应瓶内投入57.7g(0.2mol)3,3
‑
二甲氧基环丁烷
‑
1,1
‑
二羧酸二异丙酯和20%盐酸219ml混合,加入甲基三辛基氯化铵5.8g,升温至75℃,反应12小时,加入20%碳酸钠调ph=2.0
‑
2.5,加入乙酸乙酯萃取,分层,水相用乙酸乙酯萃取,合并有机相减压浓缩除去溶剂,加入80ml甲苯和40ml正庚烷重结晶得到3
‑
氧代环丁烷基羧酸19.6g,收
率85.9%,gc:99.6%。1hnmr(400mhz,cdcl3):12.15(s,1h),3.37
‑
3.28(m,5h).
[0036]
实施例5
[0037][0038]
在氮气保护下,向反应瓶内投入52.1g(0.2mol)3,3
‑
二甲氧基环丁烷
‑
1,1
‑
二羧酸二乙酯和20%盐酸219ml混合,加入甲基三辛基氯化铵5.2g,升温至75℃,反应14小时,加入20%碳酸钠调节ph=2.0
‑
2.5,加入乙酸乙酯萃取,分层,水相用乙酸乙酯萃取,合并有机相减压浓缩除去溶剂,加入80ml甲苯和40ml正庚烷重结晶得到3
‑
氧代环丁烷基羧酸18.7g,收率82.1%,gc:99.7%。
[0039]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

