(e)
‑4‑
(咪唑基甲基)肉桂酸酯的制备方法
技术领域
1.本技术属于化学制药领域,涉及一种奥扎格雷钠关键中间体(e)
‑4‑
(咪唑基甲基)肉桂酸酯的制备方法。
背景技术:
2.奥扎格雷钠(ozagrel sodium)是一种血栓素合成酶抑制剂,能阻碍前列前素h2(pgh2)生成血栓烷a2(txa2),促进pgi2的生成,增加脑血流量,抑制栓塞形成,抑制血小板凝聚,促进血栓溶解,临床用于治疗急性血栓性脑梗塞和脑梗塞所伴随的运动障碍。该品种是由日本小野药品工业株式会社及kissei药品工业株式会社共同开发,于1988年在日本获准上市,1997年在韩国上市。
3.已公开的奥扎格雷钠的合成工艺路线较少,主要的合成路线是以对甲基肉桂酸酯为起始物料,经溴化得到4
‑
溴甲基肉桂酸酯,4
‑
溴甲基肉桂酸酯进一步与咪唑反应,生成中间体(e)
‑4‑
(咪唑基甲基)肉桂酸酯,该中间体可与氢氧化钠反应生成奥扎格雷钠。具体的合成反应式如下:
[0004][0005]
目前已公开报道的专利中,该路线起始物料常用酯为对甲基肉桂酸甲酯和对甲基肉桂酸乙酯。根据《中国药典》2015版收载描述,奥扎格雷钠易溶于水。目前上市剂型有注射用奥扎格雷钠和奥扎格雷注射液等,选择合适的合成路线不仅对于收率成本有影响,对于药品质量控制尤其是注射剂的质量控制更为重要,这关乎药品安全。
[0006]
根据已公开的文献,化合物ii(e)
‑4‑
(咪唑基甲基)肉桂酸酯的合成易于产生二聚体杂质,且生成量巨大,从而导致收率降低,该杂质去除较为困难,将严重影响制剂成品的质量,因此探所制备高纯度的(e)
‑4‑
(咪唑基甲基)肉桂酸酯制备方法较为关键。
[0007][0008]
关于化合物ii(e)
‑4‑
(咪唑基甲基)肉桂酸酯的合成,已有多种技术方案寻求解决方法,提升收率,减少副反应的发生。
[0009][0010]
中国专利cn201010166699.0公开的技术方案是将溴甲基肉桂酸乙酯与咪唑、氢氧化钾在乙醚体系中进行,尝试了碱换用无水碳酸钠,溶剂换用乙醚、乙酸乙酯等方式,最终收率范围37.9%~47.1%,未能有效提高收率。
[0011]
中国专利cn201110455775.4、cn201110455771.6公开的方案是将溴甲基肉桂酸酯与咪唑、无水碳酸钾、碘化钾与丙酮混合,加热回流14~18h,过滤,滤液浓缩,残余物溶于乙酸乙酯,水洗,干燥,过滤,滤液蒸干,干燥得到固体,经乙酸乙酯/石油醚结晶,浓缩干燥得到奥扎格雷酯,专利公开的(e)
‑4‑
(咪唑基甲基)肉桂酸甲酯收率达到92.8%,(e)
‑4‑
(咪唑基甲基)肉桂酸乙酯收率达到93.5%。该方案使用昂贵的碘化钾试剂和易制毒化学品丙酮,反应加热回流14~18h时间比较长,增加了产业化的难度,由于(e)
‑4‑
(咪唑基甲基)肉桂酸酯在乙酸乙酯中的溶解度有限,因此后处理需要使用大量的乙酸乙酯,导致溶剂体积和反应规模急剧增加。且经试验验证,该体系加热后颜色变深,出现黏状物,收率明显低于预期。
[0012]
中国专利cn201110293149.x公开的方法为将naoh溶于去离子水中,搅拌下加入咪唑,搅拌混合均匀后在50
‑
80℃下反应3
‑
5h,真空检验蒸出80~90%的水分,降至室温;加入甲苯,回流分水后蒸出甲苯,降至室温后加入丙酮,得到丙酮溶液;将溴甲基肉桂酸甲酯加入丙酮溶液中,室温下反应3
‑
4h,反应完毕蒸出丙酮;残余物加入去离子水析晶,离心,干燥,得到产品,收率93.7%,纯度99.8%。该方案首先制备咪唑的钠盐,真空蒸出80~90%的水分,然后继续加入甲苯,分水后继续蒸出甲苯,再加入丙酮形成溶液,且不说如此操作是否有意义,但就该方案蒸馏水分和甲苯均为大热容和沸点较高的溶剂,能耗巨大,不符合绿色发展理念,且产业化难度较大。
[0013]
中国专利cn201410007254.6公开的技术方案分为两步,第一步制备咪唑负离子:咪唑溶解于甲苯中,搅拌加热至咪唑溶解,加入金属钠反应2
‑
4h至金属钠反应完全,得到咪唑负离子;第二步制备咪唑甲酯的制备:咪唑负离子加入丙酮配成溶液,保温15
‑
20℃,滴加溴代甲酯的丙酮溶液,20
‑
25℃反应2
‑
4h,过滤,滤液减压浓缩,浓缩物加入纯化水结晶,得到白色固体,所得产品纯度范围85%~95。该方案使用金属钠和咪唑在甲苯中反应制备咪唑负离子,金属钠性质非常活泼,在空气中极易氧化,遇水反应,反应操作环境要求高。残余的金属钠在后续加水淬灭的时候很可能导致反应燃烧。因此该方案具有较大的操作风险。
[0014]
中国专利cn201511002015.2公开的技术方案为乙腈加入反应釜中,搅拌下加入溴代甲酯、碳酸钾、咪唑得到混合液;加热回流反应3h,反应完降至室温,减压蒸出乙腈,残余物加入去离子水,搅拌析晶,过滤后干燥得到目标产物,收率91.5%,纯度99.2%。该方案经试验验证,反应体系颜色未深橘色,收率极低,大部分生成二聚体酯杂质。
[0015]
中国专利cn201511002014.8公开的技术方案为向反应釜中加入n,n
‑
二甲基甲酰胺、氢氧化钾(或氢氧化钠)、咪唑,搅拌,加入碳酸钾(或碳酸钠),10
‑
20℃搅拌,通盐水降温至0℃以下;将溴代甲酯溶于n,n
‑
二甲基甲酰胺中,滴入上述碱液中,控温15℃以下,加毕继续反应3
‑
4h,反应完毕,加入去离子水搅拌析晶得到产品,收率79.5%,纯度99.2%。该方案使用n,n
‑
二甲基甲酰胺作溶剂,反应加入了多种碱,后处理加水析晶,由于n,n
‑
二甲基甲酰
胺与水互溶,导致大量含n,n
‑
二甲基甲酰胺的废水产生,增加环境保护风险。且n,n
‑
二甲基甲酰胺含有醛基,在强碱作用下不稳定,易于发生歧化反应和分解。
[0016]
中国专利cn201711058278.4公开的技术方案为以thf为溶剂,加入咪唑和naoh,冷却至10℃,将溴代物溶于thf中,滴加入咪唑碱中,2h加毕,搅拌0.5h,控温10℃,hplc监控结束反应;后处理多次使用乙酸乙酯萃取,最后用乙酸乙酯/环己烷结晶,得到产品,收率63%,纯度99.29%。该方案加料时间长,后处理使用大量乙酸乙酯萃取,导致工作量巨大,产业化效率低。
[0017]
基于对于目前技术的分析,申请人认为,现有技术均存在缺陷,急需开发一种能降低杂质,提升收率的(e)
‑4‑
(咪唑基甲基)肉桂酸酯的合成方法。
[0018]
(e)
‑4‑
(咪唑基甲基)肉桂酸酯的合成过程中,使用咪唑作碱,过量的咪唑也会和(e)
‑4‑
(咪唑基甲基)肉桂酸酯继续反应,生成二聚体杂质。该杂质剂型较大,溶解性较大。不易去除。如何控制反应生成(e)
‑4‑
(咪唑基甲基)肉桂酸酯后不会继续反应生成杂质是一个关键问题。研究发现,单纯控制咪唑的用量无法达到减少杂质生成的目的,二者的动力学稳定性接近,取代反应具有竞争性。控制杂质的生成,主要在于控制溴甲基肉桂酸酯的反应活性。
[0019]
对于化学而言,温度是影响反应速率的关键因素之一,通过控制温度可以控制反应的选择性是一种重要的方法。
[0020]
技术实现要素:
[0021]
本技术的目的在于提供一种奥扎格雷钠关键中间体(e)
‑4‑
(咪唑基甲基)肉桂酸酯的合成方法,该方法包括以下步骤:
[0022]
(1)将咪唑、碱和溶剂加入至反应瓶中,搅拌,使混合均匀,降温;
[0023]
(2)将4
‑
溴甲基肉桂酸酯加入溶剂溶解,加入至咪唑和碱的溶液中,控温进行反应;
[0024]
(3)反应完毕,加酸淬灭并调节ph,加水析晶,过滤,洗涤,干燥,得到(e)
‑4‑
(咪唑基甲基)肉桂酸酯产品。
[0025]
步骤(1)中所述的碱选自氢氧化钠、氢氧化钾、氢氧化锂、氢化钠、氢化钙中的一种或其混合物。
[0026]
步骤(1)、步骤(2)所述溶剂选自四氢呋喃、乙腈、丙酮、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二氯甲烷中的一种或其混合物。
[0027]
步骤(1)咪唑和碱在溶剂中混合均匀后,降至
‑
10℃~10℃,优选
‑
5℃~0℃。
[0028]
步骤(2)所述4
‑
溴甲基肉桂酸酯指4
‑
溴甲基肉桂酸甲酯、4
‑
溴甲基肉桂酸乙酯。
[0029]
步骤(2)加毕4
‑
溴甲基肉桂酸酯后,反应控温
‑
10℃~10℃,优选
‑
5℃~0℃。
[0030]
步骤(3)使用稀酸调节溶液ph=6~8。
[0031]
本发明中咪唑、碱、(e)
‑4‑
(咪唑基甲基)肉桂酸酯的反应摩尔配比为1.1~2.0∶1.1~2.2∶1.0,其中咪唑、碱的摩尔比例为1.0∶1.0~1.1。
[0032]
本发明的优势在于:(1)精确掌握了(e)
‑4‑
(咪唑基甲基)肉桂酸酯合成反应控制的关键因素在于温度,通过控制温度要素,可以基本杜绝副产物的生成,大大提高了转化率。(2)反应温和,在保证反应转化率的前提下,将温度参数限定在易于控制的范围内,提升了操作的便利性和产业化的可操作性。(3)后处理使用酸中和反应体系中过量的碱,避免了产品在后处理中进一步水解的风险。(4)后处理通过蒸馏回收溶剂,利于环保并降低成本,使用水洗涤打浆可以除去过量的盐分,大大提升了产品的质量。
附图说明
[0033]
图1:(e)
‑4‑
(咪唑基甲基)肉桂酸酯的h nmr谱图;
[0034]
图2:(e)
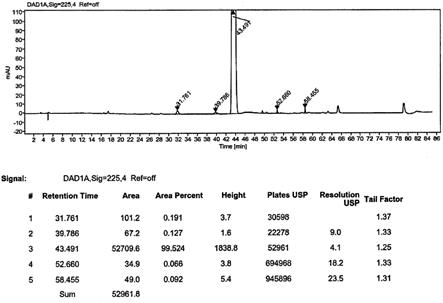
‑4‑
(咪唑基甲基)肉桂酸酯的hplc纯度谱图;
具体实施方式
[0035]
以下实施例对本发明技术方案作进一步非限制性的详细说明,其不应被认为是对本发明范围的限制,而只是本发明的示例性说明及典型代表。
[0036]
实施实例1
[0037]
于10l反应瓶中加入咪唑305g(4.5mol,1.5eq),氢氧化钠180g(4.5mol,1.5eq),四氢呋喃1.5l,常温搅拌30min,使咪唑与氢氧化钠充分反应。降温
‑
5℃~0℃继续搅拌;将4
‑
溴甲基肉桂酸甲酯765g(3.0mol,1.0eq)溶于3.0l四氢呋喃中,搅拌使溶解成溶液后,通过滴液漏斗逐步滴加至反应体系中,加毕,保持
‑
5℃~0℃继续反应2h,停止反应;加入3n盐酸调节ph至6~7,反应液减压浓缩,回收溶剂,残余物倾入20l中转桶中,继续加水10l,搅拌析晶,过滤,滤饼于60℃干燥至恒重,得白色固体,称重690g,收率95.0%。纯度99.52%。
[0038]
实施实例2
[0039]
于5l反应瓶中加入咪唑205g(3.0mol,1.2eq),氢氧化钾185g(3.3mol,1.3eq),n,n
‑
二甲基甲酰胺1.5l,常温搅拌30min,使咪唑与氢氧化钾充分反应。降温0℃~5℃继续搅拌;将4
‑
溴甲基肉桂酸甲酯635g(2.5mol,1.0eq)溶于2.0l n,n
‑
二甲基甲酰胺中,搅拌使溶解成溶液后,通过滴液漏斗逐步滴加至反应体系中,加毕,保持0℃~5℃继续反应2h,停止反应;加入3n盐酸调节ph至6~7,反应液倾入20l中转桶中,继续加水10l,搅拌析晶,过滤,滤饼于60℃干燥至恒重,得白色固体,称重550g,收率91.2%。纯度98.88%。
[0040]
实施实例3
[0041]
于10l反应瓶中加入咪唑340g(5.0mol,2.0eq),氢氧化钾280g(5.0mol,2.0eq),乙腈1.8l,常温搅拌反应1h,体系变为橘红色液,控温0℃~5℃继续搅拌反应30min;将4
‑
溴甲基肉桂酸乙酯670g(2.5mol,1.0eq)溶于3.0l乙腈中,搅拌使溶清,分批次将4
‑
溴甲基肉桂酸乙酯的乙腈溶液通过滴液漏斗逐步滴加至咪唑/氢氧化钾反应体系中,加毕,保持0℃~5℃继续反应3h,tlc监控反应(展开剂:乙酸乙酯/异丙醇/水/氨水=7ml/14ml/5ml/0.6ml,uv254nm)至原料消失停止反应;加入乙酸调节ph至6~7,反应液减压浓缩,回收乙腈,残余物倾入20l中转桶中,继续加水10l,搅拌析晶,过滤,滤饼子60℃干燥至恒重,得白色絮状固
体,称重615g,收率96.4%。纯度98.20%。
[0042]
实施实例4
[0043]
常温条件下,于100l反应釜中,加入丙酮15l,搅拌,加入咪唑3.06kg(45.0mol,1.5eq),氢氧化钠1.80kg(45.0mol,1.5eq),加毕,搅拌反应1h,降温0℃~5℃继续搅拌反应30min备用;将4
‑
溴甲基肉桂酸甲酯7.65kg(30.0mol,1.0eq)加入35l丙酮中,搅拌使溶解。分批次将4
‑
溴甲基肉桂酸甲酯的丙酮溶液滴加至咪唑/氢氧化钠反应体系中,约2h加完,加毕,继续保持0℃~5℃继续反应1h,tlc监控反应(展开剂:乙酸乙酯/异丙醇/水/氨水=7ml/14ml/5ml/0.6ml,uv254nm)至原料消失停止反应;加入3n盐酸调节ph至6~7,中和淬灭反应,反应液减压浓缩,回收丙酮,残余物加去离子水50l,搅拌析晶2h,过滤,滤饼加水洗涤一次后于60℃干燥至恒重,得白色块状固体,称重6.78kg,收率93.3%。
[0044]
实施实例5
[0045]
常温条件下,于50l反应釜中加入四氢呋喃12l,搅拌,加入咪唑2.65kg(39.0mol,1.3eq),氢氧化钾2.18kg(39.0mol,1.3eq),加毕,搅拌反应1h,降温
‑
5℃~0℃继续搅拌反应30min备用;将4
‑
溴甲基肉桂酸乙酯8.07kg(30.0mol,1.0eq)加入20l四氢呋喃中,搅拌使溶解。分批次将4
‑
溴甲基肉桂酸乙酯的四氢呋喃溶液滴加至咪唑/氢氧化钾/四氢呋喃反应体系中,约2h加完,加毕,继续保持
‑
5℃~0℃继续反应2h,tlc监控反应至原料消失,停止反应;加入乙酸调节ph=~7,中和碱并淬灭反应,反应液减压浓缩,回收四氢呋喃,浓缩残余物加去离子水30l,搅拌析晶,过滤,滤饼加水20l打浆洗涤,过滤,滤饼于60℃干燥至恒重,得淡黄色固体,称重6.84kg,收率89.1%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

