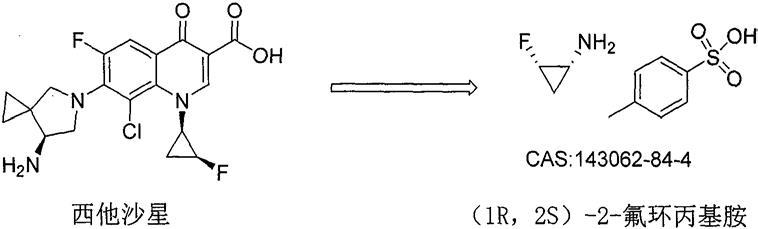
1.本发明涉及化学药物西他沙星的起始物料2-氟环丙基胺的高选择性不对称合成新方法。
背景技术:
2.西他沙星由第一三共株式会社(daiichi sankyo)研发,于2008年1月25 日获日本医药品医疗器械综合机构(pmda)批准上市,后于2019年2月1日获国家药品监督管理局(nmpa)批准上市,由第一三共株式会社在日本上市销售,商品名为西他沙星是一种氟喹诺酮类抗生素。该药适用于治疗各种感染,比如呼吸系统感染,泌尿系统感染,妇产科感染,耳鼻科感染以及牙科感染。
3.西他沙星作为一种新型的广谱喹诺酮类抗菌药,最近几年内仿制药在中国和韩国等国家也陆续上市,市场前景十分看好。随着西他沙星仿制药的上市,原料药成本控制越来越成为市场竞争的关键因素。西他沙星结构中含有的一个侧链为手性2-氟环丙基胺,该片段的合成难度大,成本高,因而导致了西他沙星原料药价格的高昂,不利于其市场推广。因此,开发新颖高效的、成本低廉的2-氟环丙基胺的合成技术势在必行。
[0004][0005]
目前合成2-氟代环丙烷甲酸的方法有如下几种。
[0006]
方法一通过使用多卤代烷烃来制备卡宾,一锅法得到环丙烷中间体。拜耳制药在1990年发表的方法中使用了丁二烯作为起始原料,将得到的环丙烷中间体上多余的烯基氧化,从而生成2-氟环丙烷甲酸(j.of fluorine chem.,1990,49,127)。
[0007][0008]
该法使用廉价的二氯一氟甲烷为起始物料时,由于其活性低,不易生成卡宾,因此环丙化反应收率低(31%);而如果使用昂贵的二溴一氟甲烷为起始物料,由于其原子利用率低,导致成本极其高昂。尤其关键的是该方法得到的产品顺反比非常差,需要经过非常繁琐的步骤才可以分开,既提高了成本,又牺牲了原子经济性。
[0009]
方法二是第一三共制药公司在1995年开发的方法,使用氟利昂与苯硫酚反应,得到的苯硫醚与丙烯酸叔丁脂反应得到相应的环丙烷中间体(jph0717945)。
[0010][0011]
该法反应的优点是获得了非常好的顺反选择性,但是得到的是对应异构体,需要进一步拆分损失一半的产物。而且生产过程中需要使用高浓度的氢氧化钾溶液和氢氧化钠溶液,并且需要加热,对设备的要求高,且会产生大量的工艺废水,不利于环保。由于反应条件剧烈,导致副反应多,产物分离必须进行精馏。而由于产物沸点很高,在工厂比较难实现精馏。
[0012]
方法三为第一三共制药公司在1996年开发的丙烯酸叔丁脂的迈克尔加成 (tetrahedron lett.1996,47,8507)。该反应在超低温下进行,使用nahmds作碱,收率为51%。得到中间体亚砜再与氟气反应得到2-氟代中间体。
[0013][0014]
该法反应的优点也是获得了非常好的顺反选择性,但是得到的是对应异构体,需要进一步拆分损失一半的产物。同时该法的在第一步使用了超低温反应,设备要求高,成本也高;而第二步使用氟气,由于氟气的强腐蚀性和氧化性,在操作性和安全性上都有着极大的问题,不适合工业化生产。
[0015]
方法四为重氮乙酸乙酯与氟代烯烃的环加成反应。卡宾与碳碳双键的加成反应是合成环丙烷的经典方法之一。第一三共在2009年发表的专利wo20100005003 中描述了使用不对称的铜催化剂,来催化1,1-氟氯乙烯与重氮乙酸乙酯的环加成反应。
[0016][0017]
该法是的优点是反应步骤比较短,而且利用手性催化剂做到了具有一定选择性的对映选择性不对称合成,但是顺反选择性仍然不好,而且其使用的1,1-氟氯烯烃为气体,在反应过程中由于氮气的释放导致其容易逃逸,使得其用量需大大过量,工艺不稳定。还有该反应需要密闭反应,导致生产上安全风险较大。
[0018]
方法五是由日本杏林制药在2014年开发的铑催化方法。该方法以方法二为基础,使用1-氟-1-苯磺酰乙烯来代替1,1-氟氯烯烃进行卡宾反应,得到的中间体中trans/cis的比例达到了86/14,大大加强了顺反选择性,采用的手性催化剂也可以达到90%以上的对映选择性。
[0019][0020]
该方法避免了使用1,1-氟氯烯烃,而是使用1-氟-1-苯磺酰乙烯,虽然避免了上述方法四的气体逃逸问题,但是1-氟-1-苯磺酰乙烯的制备困难,成本高昂 (合成路线如下):
[0021][0022]
综上所述,到目前为止,没有一条文献报道的方法在西他沙星的起始物料 2-氟环丙基胺及其前体的合成中,既可以专一性的解决顺反异构体的选择性,又可以专一的不对称合成手性中心专一的(1r,2s)-2-氟环丙基胺,严重阻碍了西他沙星在疾病治疗中的应用。
技术实现要素:
[0023]
针对现有技术存在的缺陷,本发明旨在提供一种简单、高效、低成本、适用于工业化生产的制备(1r,2s)-2-氟环丙基胺(结构式i)的方法
[0024][0025]
该合成方法既达到专一的顺反选择性,又立体专一的得到光学纯度的(1r, 2s)-2-氟环丙基胺单体。其商业化的产品甲苯磺酸盐结构如下(cas:143062-84-4)
[0026][0027]
该2-氟环丙胺的高选择性不对称合成新方法的合成路线如下式所示:
[0028][0029]
如上式路线所示,首先从化合物sma(1-氟-1苯磺酰基甲烷或者1-氟-1苯亚磺酰基甲烷)出发,在碱的作用下,在溶剂中与手性环氧氯丙烷反应生成构型专一(100%反式)的绝对构型手性取代环丙烷化合物ina,化合物ina与氧化试剂发生羟基氧化成羧基的反应得到绝对手性构型的化合物inb。化合物inb在常规curtius反应条件下发生降解反应得到绝对构型手性胺化合物inc,最后化合物 inc在金属镁在醇溶剂中或者有微量氯化汞催化的条件下脱除苯磺酰基得到专一构型手性化合物ind,ind在脱除保护基后得到化合物i,其与甲苯磺酸成盐得到商品化的化合物cas:143062-84-4。另外化合物inb也可以在金属镁在醇溶剂中或者有微量氯化汞催化的条件下脱除苯磺酰基到商品化的专一构型手性化合物 smb。
[0030]
反应第一步从化合物sma得到化合物ina反应中所用到的碱选自甲醇钠,乙醇钠,叔丁醇钾,氨基钠,lda以及hmds的锂盐,钠盐或钾盐,溶剂选自thf,乙醚以及甲基四氢呋喃。
[0031]
反应第二步从化合物ina到化合物inb反应中所用到的氧化剂选自琼斯试剂,高锰酸钾,次氯酸钠以及tempo催化的次氯酸钠。
[0032]
反应第三步从化合物inb经curtius重拍得到化合物inc中的r保护剂选自 boc,cbz。
[0033]
反应第四步从化合物inc脱除苯磺酰基得到化合物ind所用的试剂包含金属镁在甲醇,乙醇,异丙醇优选是乙醇中反应,反应过程中可以加入1%-10%当量的氯化汞来加速反应的进行。
[0034]
起始物料sma可由苯硫酚与多聚甲醛在浓盐酸的作用下得到化合物氯甲基苯硫醚ra;化合物氯甲基苯硫醚ra与csf反应得到化合物氟甲基苯硫醚rb;化合物氟甲基苯硫醚rb在氧化剂双氧水,过氧乙酸,间氯过氧苯甲酸,或高锰酸钾等试剂中发生氧化反应得到1-氟-1-苯磺酰基甲烷或者1-氟-1-苯亚磺酰基甲烷sma。具体合成路线如下:
[0035][0036]
综上所述,本发明所用的条件较已有文献报道的方法操作更加简单,物料消耗更少,且环境友好。以上优势使本发明的工艺不仅具有成本优势,更适于工业大生产,保证了西他沙星原料药的成本竞争力和持续供应。
[0037]
缩写列表
[0038]
lda
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二异丙胺锂
[0039]
tempo
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2,2,6,6-四甲基哌啶氮氧化物
[0040]
hmds
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
六甲基二硅氮烷
具体实施方式
[0041]
实施例1:1-氟-1-苯亚磺酰基甲烷(化合物4)的合成。
[0042][0043]
步骤一:合成化合物(2)。
[0044]
将多聚甲醛(45g,1.5mol)溶于甲苯(300ml)中,室温下缓慢加入浓盐酸(500ml)。反应液加热到40℃搅拌1小时,然后加入苯硫酚(110g,1mol)。反应液于60℃搅拌2小时,然后冷却至室温搅拌过夜。分离有机相,并用水(250 ml)洗两次。水相合并,并用甲苯(500ml)萃取一次。合并有机相,并用饱和食盐水(400ml)洗涤两次。有机相用无水硫酸钠干燥。经过滤,浓缩后精馏得到化合物2(128.5g,收率81%)。
[0045]
步骤二:合成化合物(3)。
[0046]
将化合物2(15.9g,0.1mol)和csf(31.9g,0.21mol)溶于peg-200/ 乙腈(1∶2,90ml)。反应液氮气保护下于80℃搅拌3小时。反应液冷却至室温,然后加入水(250ml)。溶液用氯仿(250ml)萃取两次。分离有机相,并用无水硫酸钠干燥。经过滤,减压蒸去有机溶剂得到化合物3(12.8g,收率90%)。
[0047]
步骤三:合成化合物(4)。
[0048]
将化合物3(14.2g,0.1mol)溶于二氯甲烷(200ml)中。溶液冷却至0℃,加入mcpba(75%,24.1g,0.105mol)。反应液于0℃搅拌1小时,然后加入1m氢氧化钠水溶液(150ml)。分离有机相,水相用二氯甲烷(200ml)萃取三次。合并有机相,用无水硫酸镁干燥。经过滤,加压浓缩有机溶剂得到粗品。粗品经硅胶柱纯化(石油醚∶乙酸乙酯=6∶4)得到化合物4(9.48g,收率60%)。
[0049]
实施例2:1-氟-1-苯磺酰基甲烷(化合物5)的合成。
[0050][0051]
将化合物3(14.2g,0.1mol)溶于二氯甲烷(200ml)中。溶液冷却至0℃,加入mcpba(75%,57.6g,0.25mol)。反应液升温至室温搅拌5小时,然后加入1m氢氧化钠水溶液(300ml)。分离有机相,水相用二氯甲烷(200ml)萃取三次。合并有机相,用无水硫酸镁干燥。经过滤,加压浓缩有机溶剂得到粗品。粗品经硅胶柱纯化(石油醚∶乙酸乙酯=4∶1)得到化合物5(13.9g,收率80%)。
[0052]
实施例3:(1r,2s)-2-氟环丙胺对甲苯磺酸盐(化合物11)的合成。
[0053][0054]
步骤一:合成化合物(7)。
[0055]
将化合物4(7.9g,0.05mol)和化合物6(4.6g,0.05mol)溶于thf/hmpa (13:1,140ml)中。溶液冷却至-70℃,加入lihmi)s(1m,55ml,0.055mol)。反应液于-70℃搅拌30分钟,然后加入饱和氯化铵溶液(50ml)。反应液升温至室温,加入乙酸乙酯(150ml)。分离有机相,水相用乙酸乙酯(150ml)萃取两次。合并有机相,用无水硫酸镁干燥后,经过滤,加压浓缩有机溶剂得到粗品。粗品经硅胶柱纯化(石油醚∶乙酸乙酯=6∶4)得到化合物7(7.49g,收率70%)。
[0056]
步骤二:合成化合物(8)。
[0057]
将化合物7(10.7g,0.05mol)加入甲苯(150ml)和水(200ml)的混合溶液中,然后加入高锰酸钾(23.7g,0.15mol)和四正丁基溴化铵(2.58g, 0.8mmol)。反应液于室温搅拌过夜。反应完毕后,向反应液中加入2n盐酸溶液(150ml)和乙酸乙酯(300ml)。分离有机相,水相用乙酸乙酯(300ml) 萃取两次。合并有机相,用无水硫酸钠干燥。经过滤,减压浓缩有机溶剂后得到化合物8(10.37g,收率85%)。
[0058]
步骤三:合成化合物(9)。
[0059]
将化合物8(14.64g,0.06mol)溶于叔丁醇(290ml)中,然后加入dppa (21.45g,0.078mol)和三乙胺(7.27g,0.072mol)。反应液加热回流4 小时。反应液减压浓缩除去有机溶剂,残留物溶于乙酸乙酯(300ml),分别用饱和氯化铵(150ml)、饱和碳酸氢钠(150ml)和饱和食盐水(150ml)洗涤。有机溶剂用无水硫酸镁干燥。经过滤,减压浓缩得到粗品。粗品经硅胶柱纯化(石油醚∶乙酸乙酯=4∶1)得到化合物9(12.7g,收率67%)。
[0060]
步骤三:合成化合物(10)。
[0061]
将化合物9(15.75g,0.05mol)溶于甲醇(150ml)中,于4℃加入镁粉 (3.6g,0.15mol)。反应液于室温搅拌3小时,然后倒入5%盐酸水溶液中(200 ml)。混合物用乙酸乙酯(300ml)萃取两次。合并有机相,用饱和碳酸氢钠溶液(300ml)和饱和食盐水(300ml)洗涤,然后用无水硫酸钠干燥。经过滤,减压浓缩得到粗品。粗品经硅胶柱纯化(石油醚∶乙酸乙酯=10∶1)得到化合物 10(6.91g,收率79%)。
[0062]
步骤四:合成化合物(11)。
[0063]
将化合物10(7g,0.04mol)溶于乙腈(100ml)中,加入对甲基苯磺酸 (20.64g,0.12mol)。反应液于室温搅拌24小时。减压浓缩反应液。残留物经正己烷/乙醇打浆得到化
合物11(7.7g,收率78%)。
[0064]
实施例4:(1r,2s)-2-氟-2-(苯磺酰基)环丙胺-1-羧酸(化合物8)的合成。
[0065][0066]
步骤一:合成化合物(12)。
[0067]
将化合物5(8.7g,0.05mol)和乙醇钠(20%,100ml)溶于乙醇(100ml) 中。溶液于室温搅拌30分钟,然后加入化合物6(4.6g,0.05mol)。反应液于室温搅拌过夜。减压蒸干溶剂,残留物倒入水(200ml)中。水溶液用乙酸乙酯(400ml)萃取两次。合并有机相,用无水硫酸钠干燥。经过滤,减压蒸干溶剂得到粗品。粗品经硅胶柱纯化(石油醚∶乙酸乙酯=4∶1)得到化合物12(9.5g,收率83%)。
[0068]
步骤二:合成化合物(8)。
[0069]
将化合物12(11.5g,0.05mol)加入甲苯(150ml)和水(200ml)的混合溶液中,然后加入高锰酸钾(23.7g,0.15mol)和四正丁基溴化铵(2.58 g,0.8mmol)。反应液于室温搅拌过夜。反应完毕后,向反应液中加入2n盐酸溶液(150ml)和乙酸乙酯(300ml)。分离有机相,水相用乙酸乙酯(300ml) 萃取两次。合并有机相,用无水硫酸钠干燥。经过滤,减压浓缩有机溶剂后得到化合物8(10.74g,收率88%)。
[0070]
实施例5:(1s,2s)-2-氟环丙基羧酸(化合物12)的合成。
[0071][0072]
将化合物8(5g,0.02mol)溶于甲醇(50ml)中,于4℃加入镁粉(1.92 g,0.08mol)。反应液于室温搅拌3小时,然后倒入5%盐酸水溶液中(100ml)。混合物用乙酸乙酯(200ml)萃取两次。合并有机相,用饱和食盐水(300ml) 洗涤,然后用无水硫酸钠干燥。经过滤,减压浓缩得到粗品。粗品经硅胶柱纯化 (石油醚∶乙酸乙酯=1∶1)得到化合物12(1.75g,收率84%)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

