1.本领域涉及药物化学领域,具体的涉及一种瑞德西韦的异构体及其制备和分析方法。
背景技术:
2.瑞德西韦,是由美国吉利德公司开发的一种新型实验性广谱抗病毒药物,用来针对埃博拉病毒及被认为可以有效抑制呼吸道上皮细胞中sars病毒和mers病毒。它是一种核苷酸类似前药,能有效抑制依赖rna的rna合成酶,其具体结构如下:
[0003][0004]
目前关于该药物的研究主要集中在其用途、临床试验方案、制备方法等方面,鲜有对其异构体的研究,本文旨在对其异构体进行研究,为其质控标准提供参考。
技术实现要素:
[0005]
本发明公开了一种瑞德西韦的异构体,及其制备和分析方法,其具体结构如式i所示:
[0006][0007]
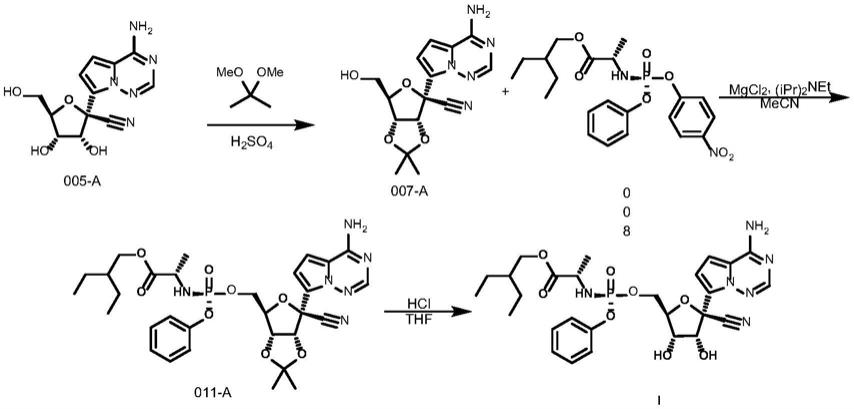
进一步的,本发明所述的式i化合物,通过005-a的化合物制备,反应路线如下:
[0008][0009]
进一步的,式i化合物制备方法如下:
[0010][0011]
具体反应步骤如下:
[0012]
(b-1)将浓硫酸滴加到(2s,3r,4s,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-二羟基-5-(羟甲基)四氢呋喃-2-腈和2,2-二甲氧基丙烷的丙酮液中;
[0013]
(b-2)将(3ar,4s,6r,6ar)-4-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-6-(羟甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3]二恶唑-4-腈,((s)-(4-硝基苯氧基)(苯氧基)磷酰基)-l-丙氨酸-2-乙基丁基酯和无水氯化镁加入到两口烧瓶中,再加入乙腈,二异丙基乙胺;
[0014]
(b-3)将((r)-((((3ar,4r,6s,6ar)-6-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-6-氰基-2,2-二甲基四氢呋喃[3,4-d][1,3]二恶唑-4-基)甲氧基)(苯氧基)磷酰基)-l-丙氨酸-2-乙基丁基酯溶于无水四氢呋喃中,然后滴加37%的浓盐酸,得式i化合物。
[0015]
进一步的,式i化合物的制备方法如下:
[0016][0017]
具体的包括以下反应步骤:
[0018]
(a-1)向4-氨基吡咯并[2,1-f][1,2,4]-三嗪的dmf溶液中加入n-碘代琥珀酰亚胺;
[0019]
(a-2)将7-碘吡咯并[2,1-f][1,2,4]三嗪-4-胺溶于四氢呋喃,滴加三甲基氯硅
烷,苯基氯化镁、异丙基氯化镁-氯化锂,再加入(3r,4r,5r)-3,4-双(苄氧基)-5-((苄氧基)甲基)二氢呋喃-2(3h)-酮的四氢呋喃溶液;
[0020]
(a)将(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-醇溶于无水二氯甲烷,滴加三甲基氰硅烷,三氟甲磺酸三甲基硅酯;
[0021]
(b)将(2r,3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-腈溶于无水二氯甲烷,加入三氯化硼,滴加甲醇,再加入三乙胺的甲醇溶液;
[0022]
(b-1)将浓硫酸滴加到(2s,3r,4s,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-二羟基-5-(羟甲基)四氢呋喃-2-腈和2,2-二甲氧基丙烷的丙酮液中;
[0023]
(b-2)将(3ar,4s,6r,6ar)-4-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-6-(羟甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3]二恶唑-4-腈,((s)-(4-硝基苯氧基)(苯氧基)磷酰基)-l-丙氨酸-2-乙基丁基酯和无水氯化镁加入到两口烧瓶中,再加入乙腈,二异丙基乙胺;
[0024]
(b-3)将((r)-((((3ar,4r,6s,6ar)-6-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-6-氰基-2,2-二甲基四氢呋喃[3,4-d][1,3]二恶唑-4-基)甲氧基)(苯氧基)磷酰基)-l-丙氨酸-2-乙基丁基酯溶于无水四氢呋喃中,然后滴加37%的浓盐酸,得式i化合物。
[0025]
进一步的,步骤(a)所述的制备方法如下:
[0026]
将(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-醇溶于无水二氯甲烷,滴加三甲基氰硅烷,三氟甲磺酸三甲基硅酯;反应结束后用碳酸氢钠水溶液淬灭,分离有机相,水相用二氯甲烷萃取,通过硅胶柱分离,用0-5%甲醇和二氯甲烷的梯度洗脱。
[0027]
进一步的,其中步骤(a)所述的0-5%甲醇和二氯甲烷的梯度洗脱条件如下:
[0028]
时间(min)梯度(甲醇:二氯甲烷)0-100:10010-500:100-5:100
[0029]
进一步的,步骤(b)的制备方法如下:将(2r,3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-腈溶于无水二氯甲烷,加入三氯化硼,滴加甲醇,再加入三乙胺的甲醇溶液;将反应混合物浓缩,二氯甲烷洗涤,保留水相,通过制备分离。
[0030]
进一步的,其中步骤(b)所述的制备分离的条件如下:
[0031]
c18反相梯度洗脱,流动相:a相:0.1%甲酸水溶液;b相:乙腈;检测波长:254nm;温度:室温;
[0032]
进一步的,其中步骤(b)所述的制备分离的条件如下:
[0033]
c18反相梯度洗脱,流动相:a相:0.1%甲酸水溶液;b相:乙腈;检测波长:254nm;温度:室温;
[0034]
具体梯度如下:
[0035]
时间(min)梯度(b%)02
101810.195129512.12142。
[0036]
进一步的,本发明提供制备式i化合物的中间体oo5-a,
[0037][0038]
进一步的,其中所述005-a的化合物通过如下步骤制备,具体反应路线如下:
[0039][0040]
将(2r,3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-腈溶于无水二氯甲烷,加入三氯化硼,滴加甲醇,再加入三乙胺的甲醇溶液;将反应混合物浓缩,二氯甲烷洗涤,保留水相,通过制备分离。
[0041]
进一步的,其中所述的制备分离的条件如下:
[0042]
c18反相梯度洗脱,流动相:a相:0.1%甲酸水溶液;b相:乙腈;检测波长:254nm;温度:室温;
[0043]
进一步的,其中所述的制备分离的条件如下:
[0044]
c18反相梯度洗脱,流动相:a相:0.1%甲酸水溶液;b相:乙腈;检测波长:254nm;温度:室温;
[0045]
具体梯度如下:
[0046]
时间(min)梯度(b%)02101810.195129512.12142。
[0047]
进一步的,005-a的制备方法如下:
[0048][0049]
具体包括以下步骤:
[0050]
(a)将(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-醇溶于无水二氯甲烷,滴加三甲基氰硅烷,三氟甲磺酸三甲基硅酯;
[0051]
(b)将(2r,3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-腈溶于无水二氯甲烷,加入三氯化硼,滴加甲醇,再加入三乙胺的甲醇溶液。
[0052]
进一步的,步骤(a)所述的制备方法如下:
[0053]
将(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-醇溶于无水二氯甲烷,滴加三甲基氰硅烷,三氟甲磺酸三甲基硅酯;反应结束后用碳酸氢钠水溶液淬灭,分离有机相,水相用二氯甲烷萃取,通过硅胶柱分离,用0-5%甲醇和二氯甲烷的梯度洗脱。
[0054]
进一步的,其中步骤(a)所述的0-5%甲醇和二氯甲烷的梯度洗脱条件如下:
[0055]
时间(min)梯度(甲醇:二氯甲烷)0-100:10010-500:100-5:100。
[0056]
进一步的,005-a的制备方法如下:
[0057]
(a)将(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-醇溶于无水二氯甲烷,滴加三甲基氰硅烷,三氟甲磺酸三甲基硅酯;反应结束后用碳酸氢钠水溶液淬灭,分离有机相,水相用二氯甲烷萃取,通过硅胶柱分离,用0-5%甲醇和二氯甲烷的梯度洗脱,梯度条件如下:
[0058]
时间(min)梯度(甲醇:二氯甲烷)0-100:10010-500:100-5:100;
[0059]
(b)将(2r,3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-腈溶于无水二氯甲烷,加入三氯化硼,滴加甲醇,再加入三乙胺的甲醇溶液;将反应混合物浓缩,二氯甲烷洗涤,保留水相,通过制备分离,c18反相梯度洗脱,流动相:a相:0.1%甲酸水溶液;b相:乙腈;检测波长:254nm;温度:室温;洗脱梯度如下:
[0060][0061][0062]
通过上述制备方法,可以快速制备纯度高的中间体005-a,通过上述中间体可以制备得到高纯度的式i化合物。
[0063]
进一步的,本发明还提供一种瑞德西韦异构体的分析方法,分析方法如下:
[0064]
通过高效液相色谱法进行正相分离,流动相:正己烷-异丙醇-三乙胺;流速:1.0ml/min;柱温:25℃;检测波长:245nm;溶剂:乙醇。
[0065]
进一步的,所述流动相,正己烷-异丙醇-三乙胺三者的体积比为500:500:1。
[0066]
进一步的,所述分析方法的色谱柱为硅胶表面共价键合有纤维素-三(3,5-二氯苯基氨基甲酸酯)为填充剂的手性柱,优选chiralpak ic色谱柱,其具体尺寸为4.6mm
×
250mm,5μm。
[0067]
上述分析方法,分离度好,能有效的分离式i化合物与瑞德西韦。
[0068]
进一步的,本发明还提供式i化合物作为瑞德西韦杂质对照品中的用途。
[0069]
某些缩写和首字母缩略词用于描述试验细节,尽管本领域技术人员可以理解其中的大部分,下表给出了其定义。
[0070]
表1缩写以及首字母缩略词的含义
[0071]
附图说明
[0072]
图1实施例4-1制备的化合物的1h nmr图;
[0073]
图2实施例7制备的化合物的1h nmr图;
[0074]
图3实施例7制备的化合物的质谱图;
[0075]
图4实施例12系统适配性溶液分析图谱;
[0076]
图5实施例12供试品溶液分析图谱。
具体实施例
[0077]
下面结合实施例对本发明的浓度分析方法的具体实施方式做进一步详细说明。这些实施方式仅用于说明本发明,而非对本发明的限制。
[0078]
化合物的hplc分析方法具体如下:
[0079]
高效液相色谱法(中国药典2015年版四部通则0512);
[0080]
色谱条件:流动相:a相:0.1%甲酸水溶液;b相:乙腈
[0081]
色谱柱:kromasil,100-5-c18,4.6
×
150mm
[0082]
梯度:
[0083]
时间(min)梯度(b%)05251295149514.15165
[0084]
检测波长:254nm
[0085]
温度:室温。
[0086]
实施例1 7-碘吡咯并[2,1-f][1,2,4]三嗪-4-胺的制备
[0087]
向4-氨基吡咯并[2,1-f][1,2,4]-三嗪(19.5g,0.145mol)的dmf(137ml)冷溶液中分5批加入n-碘代琥珀酰亚胺(33.72g),同时保持内温在0℃左右。反应完成后(在0℃下约3小时,点板监测),将反应混合物转移到1m氢氧化钠水溶液(522ml)中,将所得浆液在约常温下搅拌1.5小时,然后过滤,用水(100ml)冲洗。将固体置于50℃下真空干燥,得到30g褐色固体。
[0088]
实施例2(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-醇的制备
[0089]
将7-碘吡咯并[2,1-f][1,2,4]三嗪-4-胺(32.2g,76.9mmol)溶于四氢呋喃(480ml),在氮气保护下滴加三甲基氯硅烷(16.87g,153.8mmol,2eq.),在室温搅拌10分钟后,置于冰水浴中。待冷却至约0℃时,缓慢滴加苯基氯化镁(2m,77ml,2eq.)。加料完毕后继续搅拌20分钟,然后缓慢滴加异丙基氯化镁-氯化锂(1.3m,62ml,1.05eq.),滴加过程中保持内部温度低于5℃。滴加完毕后,将反应混合物冷却至约-20℃,并加入(3r,4r,5r)-3,4-双(苄氧基)-5-((苄氧基)甲基)二氢呋喃-2(3h)-酮(20g,76.9mmol,1eq.)的四氢呋喃(97ml)溶液,同时保持内部反应温度约为-20℃。加料完毕后继续反应1小时,然后升温至0℃,然后用甲醇(64ml)淬灭,随后加入乙酸(64ml)和水(64ml),将所得混合物升温至室温,然后减压浓缩。将得到的浓缩物在乙酸乙酯(800ml)和盐酸水溶液(1m,800ml)之间分配。分
离有机层,分别用饱和碳酸氢钠水溶液(800ml)和饱和食盐水(800ml)洗涤,用无水硫酸钠干燥,减压浓缩。通过硅胶柱分离,用0-5%甲醇和二氯甲烷的梯度洗脱,得到17.6g淡黄色固体。
[0090]
实施例3(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-腈的制备
[0091]
实施例3-1
[0092]
将(3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-醇(8.0g,14.48mmol)溶于无水二氯甲烷(320ml)中并在氮气保护下置于冰水浴中搅拌,当内温降到2℃左右时,滴加三甲基氰硅烷(7.18g,72.4mmol,5eq.)。加料完毕后再搅拌10分钟,然后滴加三氟甲磺酸三甲基硅酯(16.14g,72.4mmol,5eq.),控制内部温度在5℃以下。继续在冰水浴中反应2小时。反应结束后加入300ml饱和碳酸氢钠水溶液淬灭。反应混合物继续搅拌10分钟后,分离有机相,水相用二氯甲烷萃取三次(100ml
×
3),收集有机相用无水硫酸钠干燥后,减压浓缩。通过硅胶柱分离,用0-5%甲醇和二氯甲烷的梯度洗脱,具体梯度洗脱条件如下,
[0093]
时间(min)梯度(甲醇:二氯甲烷)0-100:10010-500:100-5:100
[0094]
最终得到6g淡黄色固体。
[0095]
实施例4(2s,3r,4s,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-二羟基-5-(羟甲基)四氢呋喃-2-腈的制备
[0096]
实施例4-1
[0097]
将(2r,3r,4r,5r)-2-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-3,4-双(苄氧基)-5-((苄氧基)甲基)四氢呋喃-2-腈(6.0g,10.7mmol)溶于无水二氯甲烷(60ml)中并在氮气保护下冷却至-78℃。然后加入三氯化硼(1m,40.6ml,3.8eq.),将反应混合物升温至-40℃,搅拌2小时。然后将反应混合物再冷却至-78℃,并滴加甲醇(12ml)。滴加三乙胺(16ml)的甲醇(24ml)溶液,最后将反应混合物升温至室温。将混合物浓缩,剩余固体残余物用甲醇(60ml)溶解,并加热至45℃。加入水(60ml),并将得到的混合物在45℃减压浓缩,以除去挥发物,然后用二氯甲烷洗三遍(30ml
×
3),保留水相,通过制备分离,制备液相具体条件如下:
[0098]
色谱条件:流动相:a相:0.1%甲酸水溶液;b相:乙腈
[0099]
色谱柱:kromasil,100-5-c18,30
×
150mm
[0100]
梯度:
[0101]
时间(min)梯度(b%)02101810.195129512.12142
氰基-2,2-二甲基四氢呋喃[3,4-d][1,3]二恶唑-4-基)甲氧基)(苯氧基)磷酰基)-l-丙氨酸-2-乙基丁基酯(490mg,0.76mmol)溶于无水四氢呋喃中(3.8ml),在冰水浴中搅拌10分钟,然后滴加浓盐酸(0.76ml)。在常温下搅拌反应5小时。然后加入3.8ml水,用饱和碳酸钠水溶液调节ph至8。混合物用乙酸乙酯(5ml
×
3)萃取,有机相用无水硫酸钠干燥后浓缩后,通过硅胶柱分离,用0-10%甲醇和二氯甲烷的梯度洗脱,得到250mg白色粉末状固体,纯度为93.33%。
[0116]1h nmr(400mhz,dmso-d6)δ7.91(s,3h),7.37(t,j=7.9hz,2h),7.25(d,j=8.4hz,2h),7.18(t,j=7.3hz,1h),6.91
–
6.87(m,1h),6.64(d,j=4.4hz,1h),6.04(dd,j=13.0,10.1hz,1h),4.77(d,j=4.1hz,1h),4.36(dt,j=11.7,5.8hz,2h),4.22
–
4.16(m,1h),4.11(dt,j=12.4,6.3hz,1h),4.00(dd,j=10.9,5.9hz,1h),3.93(dt,j=10.0,6.8hz,2h),1.51
–
1.41(m,1h),1.38
–
1.21(m,10h),0.82(td,j=7.4,2.4hz,6h)(详见附图2)。lcms:[m h] :603.3(详见附图3)。
13
c nmr(101mhz,dmso-d6)δ173.71,173.65,155.91,151.29,151.23,148.31,139.67,130.04,125.38,124.98,122.95,120.73,120.69,118.42,116.02,110.28,101.22,81.22,81.15,78.21,75.71,71.86,66.63,66.43,50.25,34.86,30.89,23.06,23.02,21.51,20.36,20.30,11.27,11.22。
[0117]
实施例8瑞德西韦异构体分析方法
[0118]
高效液相色谱法(中国药典2015年版四部通则0512):
[0119]
系统适用性溶液配置:取瑞德西韦及对映体各适量,加乙醇超声溶解并稀释制成每1ml中约含瑞德西韦0.5mg和对映体2.5μg的混合溶液;
[0120]
色谱柱:chiralpak od-h(4.6mm
×
250mm,5μm)
[0121]
流动相:正己烷-异丙醇(80:20)
[0122]
流速:1.0ml/min
[0123]
柱温:25℃
[0124]
检测波长:245nm
[0125]
进样量:20μl
[0126]
取系统适用性溶液注入液相色谱,结果:异构体与瑞德西韦保留时间重合,无法分离。
[0127]
实施例9瑞德西韦异构体分析方法
[0128]
高效液相色谱法(中国药典2015年版四部通则0512):
[0129]
系统适用性溶液配置:取瑞德西韦及对映体各适量,加乙醇超声溶解并稀释制成每1ml中约含瑞德西韦0.5mg和对映体2.5μg的混合溶液;
[0130]
色谱柱:chiralpak ia(4.6mm
×
250mm,5μm)
[0131]
流动相:正己烷-异丙醇(60:40)
[0132]
流速:1.0ml/min
[0133]
柱温:25℃
[0134]
检测波长:245nm
[0135]
进样量:20μl
[0136]
取系统适用性溶液注入液相色谱,结果:瑞德西韦峰拖尾严重、异构体在其拖尾峰上。
[0137]
实施例10瑞德西韦异构体分析方法
[0138]
高效液相色谱法(中国药典2015年版四部通则0512):
[0139]
系统适用性溶液配置:取瑞德西韦及对映体各适量,加乙醇超声溶解并稀释制成每1ml中约含瑞德西韦0.5mg和对映体2.5μg的混合溶液;
[0140]
色谱柱:chiralpak ic(4.6mm
×
250mm,5μm)
[0141]
流动相:正己烷-异丙醇-三氟乙酸(500:500:1)
[0142]
流速:1.0ml/min
[0143]
柱温:25℃
[0144]
检测波长:245nm
[0145]
进样量:20μl
[0146]
取系统适用性溶液注入液相色谱,结果:瑞德西韦峰峰扩散严重、灵敏度较低。
[0147]
实施例11瑞德西韦异构体分析方法
[0148]
高效液相色谱法(中国药典2015年版四部通则0512):
[0149]
系统适用性溶液配置:取瑞德西韦及对映体各适量,加乙醇超声溶解并稀释制成每1ml中约含瑞德西韦0.5mg和对映体2.5μg的混合溶液;
[0150]
流动相:正己烷-异丙醇-三乙胺(500:500:1)
[0151]
流速:1.0ml/min
[0152]
柱温:25℃
[0153]
检测波长:245nm
[0154]
进样量:20μl
[0155]
取系统适用性溶液注入液相色谱,结果:能分离,但是峰对称性略差。
[0156]
实施例12瑞德西韦异构体分析方法
[0157]
高效液相色谱法(中国药典2015年版四部通则0512):
[0158]
系统适用性溶液配置:取瑞德西韦及对映体各适量,加乙醇超声溶解并稀释制成每1ml中约含瑞德西韦0.5mg和对映体2.5μg的混合溶液;
[0159]
供试品溶液:取本品适量,加乙醇超声溶解并稀释制成每1ml中约含0.5mg的溶液,即得。
[0160]
流动相:正己烷-异丙醇-三乙胺(500:500:2)
[0161]
流速:1.0ml/min
[0162]
柱温:25℃
[0163]
检测波长:245nm
[0164]
进样量:20μl
[0165]
将系统适配性溶液以及供试品溶液注入色谱仪,结果:分离度良好,且峰型佳,系统适配性溶液图谱详见附图4,其中式i化合物保留时间为5.084,瑞德西韦保留时间为8.485,供试品溶液详见附图5。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

