1.本发明涉及液晶显示材料领域,具体涉及具有负介电各向异性的液晶化合物、液晶组合物及液晶显示器件。
背景技术:
2.目前,液晶化合物的应用范围拓展的越来越广,其可应用于多种类型的显示器、电光器件、传感器等中。用于上述显示领域的液晶化合物的种类繁多,其中向列相液晶应用最为广泛。向列相液晶已经应用在无源tn、stn矩阵显示器和具有tft有源矩阵的系统中。
3.对于薄膜晶体管技术(tft
‑
lcd)应用领域,近年来市场虽然已经非常巨大,技术也逐渐成熟,但人们对显示技术的要求也在不断的提高。随着tft
‑
lcd的不断发展,宽视角模式已成为行业内追求的目标,目前主流的宽视角技术主要采用va垂直取向、ips面内开关及ffs边缘场开关等显示类型。这些显示模式,广泛采用具有负介电各向异性的液晶介质。
4.对于用于这些模式的液晶介质,对其响应时间的要求越来越高。而液晶介质的响应时间受到清亮点t
ni
(℃)、折光率(δn)、介电常数(δε)、弹性系数(kii、pn)、旋转粘度(gamma 1,mpa.s)等多个因素的影响,如何获得这些因素综合作用下的响应时间提高的液晶化合物是本领域亟待解决的问题之一。
技术实现要素:
5.本发明针对上述现有技术存在的问题,进行了深入的研究后发现,采用本发明的式i所示的液晶化合物,能够获得在维持一定水平的负型介电常数的基础上具有提高的响应时间的新型液晶化合物,由此完成了本发明。
6.对于液晶介质,根据显示模式的不同,液晶介质的响应时间与g1/(k
11
*
△
n*
△
n)或者g1/(k
33
*
△
n*
△
n)相关。具体地,在va(vertical alignment,垂直取向)或者ps
‑
va(polymer stabilized vertical alignment,聚合物稳定垂直取向)模式下,液晶介质的响应时间与g1/(k
33
*
△
n*
△
n)的值相关,而在ffs(fringe field switching,边缘场开关)、ips(in
‑
plane switching,平面转换)、ps
‑
ffs(polymer stabilized fringe field switching,聚合物稳定边缘场开关)、ps
‑
ips(polymer stabilized in
‑
plane switching,聚合物稳定平面转换)等模式下,液晶介质的响应时间与g1/(k
11
*
△
n*
△
n)的值相关。
7.在本技术中,将g1/(k
33
*
△
n*
△
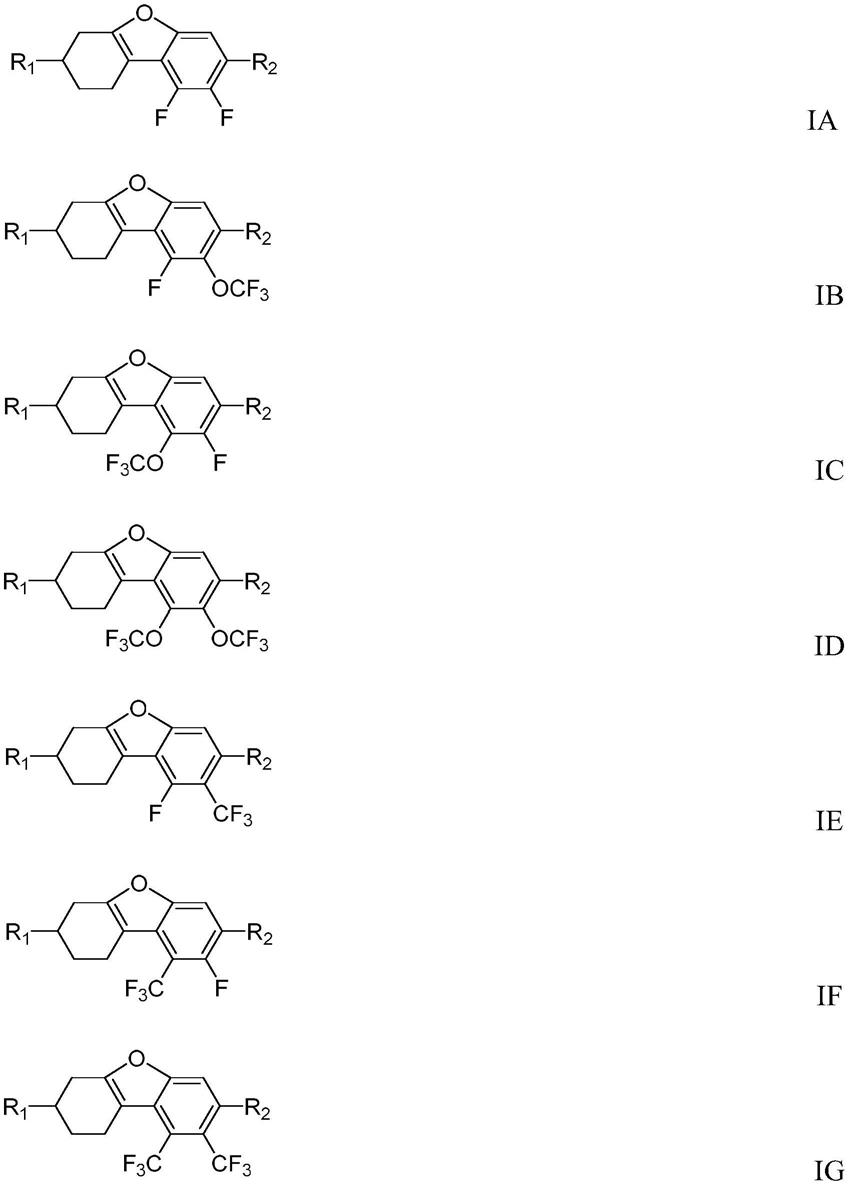
n)、g1/(k
11
*
△
n*
△
n)的值称为响应指标值。前述响应指标值越小,表明液晶介质的响应时间越快。
8.本发明的具有负介电各向异性的液晶化合物在维持一定水平的负型介电常数的基础上具有小的响应指标值从而具有提高的响应时间。
9.本发明包括下述的技术方案:
10.一方面,本发明提供一种具有负介电各向异性的液晶化合物,所述化合物具有下述的式ⅰ所示的结构:
[0011][0012]
式i中,r1、r2各自独立地表示氢原子、碳原子数为1~8的直链烷基、碳原子数为1~8的直链烷氧基、碳原子数为2~8的直链烯基、碳原子数为2~8的直链烯氧基,其中一个或两个不相邻的
‑
ch2‑
任选被
‑
o
‑
取代,任意h任选被f原子取代;
[0013]
环a1选自下述的基团组成的组:1,4
‑
亚环己基、环己烯
‑
1,4
‑
二基、1,4
‑
亚苯基、2
‑
氟
‑
1,4
‑
亚苯基、2,3
‑
二氟
‑
1,4
‑
亚苯基、氧杂环己烷
‑
2,5
‑
二基、1,3
‑
二氧杂环己烷
‑
2,5
‑
二基、1
‑
甲基环己烷
‑
1,4
‑
二基、2
‑
甲基环己烷
‑
1,4
‑
二基、2
‑
甲基苯
‑
1,4
‑
二基;
[0014]
z表示单键、
‑
c2h2‑
、
‑
c2h4‑
、
‑
c2h2ch2o
‑
、
‑
och2c2h2‑
、
‑
ch2o
‑
、
‑
och2‑
、
‑
c2h2ch2s
‑
、
‑
sch2c2h2‑
、
‑
ch2s
‑
、
‑
sch2‑
、
‑
o
‑
、
‑
s
‑
、
‑
cf2o
‑
、
‑
ocf2‑
、
‑
c≡c
‑
、
‑
ooc
‑
、或者
‑
coo
‑
,其中
‑
ch2o
‑
、
‑
c2h2‑
、
‑
c2h4‑
、
‑
c2h2ch2o
‑
、
‑
och2c2h2‑
中的任意h任选被f取代;
[0015]
x表示
‑
o
‑
、
‑
s
‑
、
‑
so
‑
、
‑
soo
‑
、
‑
cf2‑
、
‑
co
‑
或者
‑
ch2‑
;
[0016]
y1、y2各自独立地表示
‑
f
‑
、
‑
ch2f
‑
、
‑
chf2‑
、
‑
cf3‑
、
‑
och2f
‑
、
‑
ochf2‑
或者
‑
ocf3‑
;
[0017]
n表示0、1、2或者3。
[0018]
本发明另一方面提供一种液晶组合物,其含有前述的本发明的具有负介电各向异性的液晶化合物。
[0019]
本发明的又一方面提供一种液晶显示器件,其含有前述的本发明的具有负介电各向异性的液晶化合物或者前述的本发明的液晶组合物。
[0020]
发明效果
[0021]
与现有技术相比,本发明的具有负介电各向异性的液晶化合物在维持一定水平的负型介电常数的基础上具有小的响应指标值从而具有更快的响应时间。通过在本发明的液晶组合物中使用本发明的具有负介电各向异性的液晶化合物,在本发明的液晶显示器件中含有使用了本发明的液晶化合物的液晶组合物,能够使得显示装置的响应时间更快。
附图说明
[0022]
图1为本发明的实施例1中制备的化合物loy
‑3‑
o2溶于cdcl3的1h核磁共振光谱图。
[0023]
图2为本发明的实施例1中制备的化合物loy
‑3‑
o2溶于cdcl3的
13
c核磁共振光谱图。
具体实施方式
[0024]
以下将结合具体实施方案来说明本发明。需要说明的是,下面的实施例为本发明的示例,仅用来说明本发明,而不用来限制本发明。在不偏离本发明主旨或范围的情况下,可进行本发明构思内的其他组合和各种改良。
[0025]
[具有负介电各向异性的液晶化合物]
[0026]
本发明的具有负介电各向异性的液晶化合物具有下述的式ⅰ所示的结构:
[0027][0028]
式i中,r1、r2各自独立地表示氢原子、碳原子数为1~8的直链烷基、碳原子数为1~8的直链烷氧基、碳原子数为2~8的直链烯基、或者碳原子数为2~8的直链烯氧基,其中一个或两个不相邻的
‑
ch2‑
任选被
‑
o
‑
取代,任意h任选被f原子取代。
[0029]
环a1选自下述的基团组成的组:1,4
‑
亚环己基、环己烯
‑
1,4
‑
二基、1,4
‑
亚苯基、2
‑
氟
‑
1,4
‑
亚苯基、2,3
‑
二氟
‑
1,4
‑
亚苯基、氧杂环己烷
‑
2,5
‑
二基、1,3
‑
二氧杂环己烷
‑
2,5
‑
二基、1
‑
甲基环己烷
‑
1,4
‑
二基、2
‑
甲基环己烷
‑
1,4
‑
二基、2
‑
甲基苯
‑
1,4
‑
二基。
[0030]
前述的2
‑
氟
‑
1,4
‑
亚苯基表示下述的2个二价基团,其中的氟取代基可以位于左侧,也可以位于右侧。其他类似的基团也适用该规则。
[0031][0032]
z表示单键、
‑
c2h2‑
、
‑
c2h4‑
、
‑
c2h2ch2o
‑
、
‑
och2c2h2‑
、
‑
ch2o
‑
、
‑
och2‑
、
‑
c2h2ch2s
‑
、
‑
sch2c2h2‑
、
‑
ch2s
‑
、
‑
sch2‑
、
‑
o
‑
、
‑
s
‑
、
‑
cf2o
‑
、
‑
ocf2‑
、
‑
c≡c
‑
、
‑
ooc
‑
或者
‑
coo
‑
,其中
‑
ch2o
‑
、
‑
c2h2‑
、
‑
c2h4‑
、
‑
c2h2ch2o
‑
、
‑
och2c2h2‑
中任意h任选被f取代;
[0033]
x表示
‑
o
‑
、
‑
s
‑
、
‑
so
‑
、
‑
soo
‑
、
‑
cf2‑
、
‑
co
‑
或者
‑
ch2‑
;
[0034]
y1、y2各自独立地表示
‑
f
‑
、
‑
ch2f
‑
、
‑
chf2‑
、
‑
cf3‑
、
‑
och2f
‑
、
‑
ochf2‑
或者
‑
ocf3‑
;
[0035]
n表示0、1、2或3。
[0036]
作为前述的“碳原子数为1~8的直链烷基”,可以列举出例如,甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基等。
[0037]
作为前述的“碳原子数为1~8的直链烷氧基”,可以列举出例如,甲氧基、乙氧基、正丙氧基、正丁氧基、正戊氧基、正己氧基、正庚氧基、正辛氧基等。
[0038]
作为前述的“碳原子数为2~8的直链烯基”,可以列举出例如,乙烯基、1
‑
丙烯基、2
‑
丙烯基、1
‑
丁烯基、2
‑
丁烯基、3
‑
丁烯基、1
‑
戊烯基、2
‑
戊烯基、1
‑
己烯基、2
‑
己烯基、3
‑
己烯基、1
‑
庚烯基、2
‑
庚烯基、3
‑
庚烯基、1
‑
辛烯基、2
‑
辛烯基、3
‑
辛烯基。
[0039]
作为前述的“碳原子数为2~8的直链烯氧基”,可以列举出例如,乙烯氧基、1
‑
丙烯氧基、2
‑
丙烯氧基、1
‑
丁烯氧基、2
‑
丁烯氧基、3
‑
丁烯氧基、1
‑
戊烯氧基、2
‑
戊烯氧基、1
‑
己烯氧基、2
‑
己烯氧基、3
‑
己烯氧基、1
‑
庚烯氧基、2
‑
庚烯氧基、3
‑
庚烯氧基、1
‑
辛烯氧基、2
‑
辛烯氧基、3
‑
辛烯氧基等。
[0040]
前述的“一个或两个不相邻的
‑
ch2‑
任选被
‑
o
‑
取代”是指,前述的碳原子数为1~8的直链烷基、碳原子数为1~8的直链烷氧基、碳原子数为2~8的直链烯基、碳原子数为2~8的直链烯氧基中的任意
‑
ch2‑
任选被取代为
‑
o
‑
,但是相邻的
‑
ch2‑
不会同时被取代。
[0041]
前述的“任意h任选被f原子取代”,是指,对于f取代的个数没有任何的限定,可以为单氟取代、多氟取代、或者全氟取代。
[0042]
优选地,前述r1表示氢原子、碳原子数1~5的直链烷基、碳原子数1~5的直链烷氧
基、碳原子数2~5的直链烯基、或者、碳原子数2~5的直链烯氧基,其中一个或两个不相邻的
‑
ch2‑
任选被
‑
o
‑
取代,任意h任选被f原子取代。
[0043]
前述的“碳原子数1~5的直链烷基”,可以列举出例如,甲基、乙基、正丙基、正丁基、正戊基等。优选为甲基、乙基或者正丙基。
[0044]
作为前述的“碳原子数1~5的直链烷氧基”,可以列举出例如,甲氧基、乙氧基、正丙氧基、正丁氧基、正戊氧基。优选为甲氧基、乙氧基或者正丙氧基。
[0045]
作为前述的“碳原子数2~5的直链烯基”,可以列举出例如,乙烯基、1
‑
丙烯基、2
‑
丙烯基、1
‑
丁烯基、2
‑
丁烯基、3
‑
丁烯基、1
‑
戊烯基、2
‑
戊烯基、3
‑
戊烯基。优选为乙烯基、1
‑
丙烯基、3
‑
丁烯基、或者、3
‑
戊烯基。
[0046]
作为前述的“碳原子数2~5的直链烯氧基”,可以列举出例如,乙烯氧基、1
‑
丙烯氧基、2
‑
丙烯氧基、1
‑
丁烯氧基、2
‑
丁烯氧基、3
‑
丁烯氧基、1
‑
戊烯氧基、2
‑
戊烯氧基、3
‑
戊烯氧基。优选为乙烯氧基、1
‑
丙烯氧基、3
‑
丁烯氧基、或者、3
‑
戊烯氧基。
[0047]
前述的碳原子数1~5的直链烷基、碳原子数1~5的直链烷氧基、碳原子数2~5的直链烯基、或者、碳原子数2~5的直链烯氧基中,一个或两个不相邻的
‑
ch2‑
任选被
‑
o
‑
取代,任意h任选被f原子取代。
[0048]
本发明的式i所示的化合物的一些实施方式中,前述的r1优选为碳原子数1~5的直链烷基或者碳原子数2~5的直链烯基。
[0049]
本发明的式i所示的化合物的一些实施方式中,前述的r2优选为碳原子数1~5的直链烷氧基或者碳原子数2~5的直链烯氧基。
[0050]
本发明的式i所示的化合物的一些实施方式中,环a1优选为1,4
‑
亚环己基、环己烯
‑
1,4
‑
二基或者1,4
‑
亚苯基,更优选为1,4
‑
亚环己基。
[0051]
本发明的式i所示的化合物的一些实施方式中,z优选表示单键、
‑
c2h2‑
、或者
‑
c2h4‑
,更优选为单键。
[0052]
本发明的式i所示的化合物的一些实施方式中,x优选为
‑
o
‑
或者
‑
s
‑
。
[0053]
式i中,n表示0、1、2、或者3,从获得更小的响应指标值从而具有更快的响应时间等方面考虑,n优选为0、1或者2,进一步优选为n=0或者1。
[0054]
本发明的具有负介电各向异性的液晶化合物中,优选地,其选自下述的式ia~in、ia
‑
in所示化合物组成。
[0055]
[0056]
[0057]
[0058][0059]
其中,r1、r2的定义与前述相同。
[0060]
进一步,本发明的具有负介电各向异性的液晶化合物优选为选自下述的式ia
‑
1~
in
‑
4、ia
‑
1~in
‑
4所示的化合物组成的组,其中,alkyl各自独立地表示碳原子数为1~8的直链烷基、alkenyl各自独立地表示碳原子数为2~8的直链烯基,
[0061]
[0062]
[0063]
[0064]
[0065]
[0066]
[0067]
[0068]
[0069]
[0070]
[0071]
[0072]
[0073]
[0074]
[0075][0076]
作为前述的alkyl所表示的碳原子数为1~8的直链烷基,可以列举出甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基,优选为甲基、乙基或者正丙基。
[0077]
作为前述的alkenyl所表示的碳原子数为2~8的直链烯基,可以列举出例如乙烯基、1
‑
丙烯基、2
‑
丙烯基、1
‑
丁烯基、2
‑
丁烯基、3
‑
丁烯基、1
‑
戊烯基、2
‑
戊烯基、1
‑
己烯基、2
‑
己烯基、3
‑
己烯基、1
‑
庚烯基、2
‑
庚烯基、3
‑
庚烯基、1
‑
辛烯基、2
‑
辛烯基、3
‑
辛烯基等,优选为乙烯基、1
‑
丙烯基或者2
‑
丙烯基。
[0078]
[液晶化合物的制备方法]
[0079]
下面,对本发明的具有负介电各向异性的液晶化合物的制备方法进行说明。
[0080]
需要理解的是,本发明的具有负介电各向异性的液晶化合物的制备方法,并非限于下述说明的制备方法。本领域技术人员可以采用其他的适宜的方法进行制备。
[0081]
另外,下述的说明中对于式i所示的部分化合物进行说明,对于其他的化合物,本领域技术人员可以参照下述的说明并结合本领域的常规技术手段来获得。
[0082]
采用包括如下制备步骤的方法制备下述结构式所示的液晶化合物,其中,r1、r2、y1以及y2的定义与前述相同。
[0083][0084]
步骤a1:将y1以及y2取代的苯与带有r1取代基的环己酮在钯金属催化剂的存在下发生碳
‑
芳基化反应,得到苯基环己酮化合物(a1);
[0085][0086]
步骤a2:将前述的苯基环己酮化合物(a1)进一步发生钯金属催化氧
‑
芳基化反应,得到苯并呋喃化合物(b1);
[0087][0088]
步骤a3:将前述步骤a2获得的苯并呋喃化合物(b1)依次在强碱的作用下与硼酸三甲酯反应,接着进行水解和氧化反应,生成带有r1取代基以及酚羟基的苯并呋喃化合物(c1);
[0089][0090]
步骤a4:将带有酚羟基的单边r1取代的苯并呋喃化合物(c1)与r2x(x表示溴或碘)于碱性条件下反应生成两边对称或不对称的苯并呋喃化合物(i
‑
1)。
[0091][0092]
采用包括如下制备步骤的方法制备前述式
ⅰ‑
2所示的液晶化合物,其中,r1、r2、y1以及y2的定义与前述相同。
[0093][0094]
步骤b1:将y1以及y2取代的苯与带有r1取代基的环己酮进行钯金属碳
‑
芳基化反应,得到苯基环己酮化合物(a2);
[0095][0096]
步骤b2:将前述的苯基环己酮化合物(a2)与硫化试剂反应,生成对应结构的苯基环己硫酮化合物(b2);
[0097]
[0098]
步骤b3:使苯基环己硫酮化合物(b2)进行钯金属催化硫
‑
芳基化反应,获得苯并噻吩化合物(c2);
[0099][0100]
步骤b4:将带有r1取代基的苯并噻吩化合物(c2)依序使用强碱与硼酸三甲酯反应后,接着进行水解以及氧化反应,生成带有r1取代基以及酚羟基的苯并噻吩化合物(d2);
[0101][0102]
步骤b5:将带有酚羟基的单边r1基取代的苯并噻吩化合物(d2)与带有r2x(x表示溴或碘)于碱性条件反应生成两边对称或不对称的苯并呋喃化合物(i
‑
2)。
[0103][0104]
以上,示出了前述的式i
‑
1、式i
‑
2所示化合物的制备方法。对于其他化合物的制备,本领域技术人员能够参照前述制备方法,根据本领域的技术常识,改变前述制备方法中的反应原料进行制备,没有特别的限定。
[0105]
[液晶组合物]
[0106]
本发明的液晶组合物中含有前述的本发明的具有负介电各向异性的液晶化合物。
[0107]
本发明的液晶组合物中,可以含有一种或者多种本发明的负介电各向异性的液晶化合物,对其含量没有特别的限定。
[0108]
本发明的液晶组合物中,本发明的负介电各向异性的液晶化合物的含量按照重量百分含量计算可以为例如20%以下。从低温溶解性、可靠性等方面考虑,优选为15%以下的范围。含有多种本发明的负介电各向异性的液晶化合物时,本发明的负介电各向异性的液晶化合物的含量的总和以重量百分含量计算可以为例如50%以下。
[0109]
本发明的液晶组合物中,除了前述的具有负介电各向异性的液晶化合物之外,本领域技术人员还可以在不破坏其期望的液晶组合物的性能的基础上添加其他液晶化合物。
[0110]
本发明的液晶组合物中,可选的,还可以加入各种功能的掺杂剂,这些掺杂剂可以列举出例如抗氧化剂、紫外线吸收剂、手性剂。
[0111]
如前所述,本发明的液晶组合物中虽然含有本发明的具有负性介电各向异性的液晶化合物,但是本发明的组合物并非一定为负性介电各向异性,其也可以为正性介电各向异性。本领域技术人员能够根据需要调节组合物各组分的组成及配比来获得具有需要的各向异性的组合物。
[0112]
对于本发明的液晶组合物的制备,没有特别的限定。在含有本发明的液晶化合物的基础上,本领域技术人员能够根据需要,选择适宜的其他组分进行调制。
[0113]
[液晶显示器件]
[0114]
本发明的第三方面提供一种液晶显示器件,其只要包含前述的本发明的具有负介电各向异性的液晶化合物,或者前述的液晶组合物,就没有特别的限定。本发明的液晶显示器件可以为有源矩阵显示器件,也可以为无源矩阵显示器件。本领域技术人员能够根据所需的性能选择合适的液晶显示组件、液晶显示器的结构。
[0115]
实施例
[0116]
实施例1
[0117]
loy
‑3‑
o2
[0118][0119]
合成路线:
[0120][0121]
在反应瓶中加入1.13g碳酸铯(3.45mmol)、7.2mg pd2(dba)3(0.008mmol)和11mg xantphos(0.019mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽),之后将反应瓶气体置换成氮气,加入4ml无水二噁烷、0.5g 1
‑
溴
‑
3,4
‑
二氟
‑2‑
碘苯(1.57mmol)和0.44g 3
‑
丙基环己酮(3.14mmol)。接着在80℃油浴反应24小时。待反应液冷却后,用乙醚和水进行萃取,收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a3 0.41g。
[0122]1h
‑
nmr(500mhz,cdcl3,ppm):7.13(d,1h)、6.66(d,1h)、3.50(t,1h)、2.31(m,2h)、2.21(m,2h)、1.86(m,2h)、1.79(m,1h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0123]
取0.5g化合物a3(1.51mmol)、0.69g碳酸铯(2.11mmol)、34.6mg pd2(dba)3(0.038mmol)及40.7mg dpephos(0.076mmol,(2
‑
二苯基膦苯基)醚)于反应瓶内,之后将反应瓶气体置换成氮气。接着加入4ml无水甲苯后,在100℃油浴反应20小时,待冷却后将反应液以硅藻土过滤、减压浓缩,将浓缩液以柱层析纯化,可得到产物b3 0.36g。
[0124]1h
‑
nmr(500mhz,cdcl3,ppm):7.17(d,1h)、6.88(d,1h)、2.60(m,2h)、2.48(m,2h)、1.86(m,1h)、1.71(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0125]
取0.5g化合物b3(2.0mmol)溶解在12ml thf中,将反应瓶降温至
‑
78℃后加入1.4ml的含1.5m n
‑
丁基锂(2.1mmol)的己烷溶液,之后降温至0℃反应30分钟。接着在
‑
78℃下加入0.22g硼酸三甲酯(2.1mmol)后,将温度升到室温反应1小时。之后加入1ml醋酸以及
0.25ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物c3 0.45g。
[0126]1h
‑
nmr(500mhz,cdcl3,ppm):6.64(s,1h)、5.0(s,1h)、2.60(m,2h)、2.48(m,2h)、1.86(m,1h)、1.71(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0127]
取0.5g化合物c3(1.88mmol)、0.52g碳酸钾(3.76mmol)及0.35g碘乙烷(2.26mmol)于反应瓶内,加入6ml二甲基甲酰胺,在70℃油浴反应3小时,待反应液降温后以水及乙酸乙酯进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物d3 0.56g。
[0128]
制备得到的化合物d3(loy
‑3‑
o2)的质谱、核磁共振谱数据如下。根据测试结果,化合物d3为loy
‑3‑
o2所示结构。
[0129]
ms(ei,m/z):206,265,294。
[0130]1h
‑
nmr(500mhz,cdcl3,ppm):6.68(d,1h)、3.98(q,2h)、2.6(m,2h)、2.48(m,2h)、1.86(m,1h)、1.71(m,2h)、1.33
‑
1.25(m,7h)、0.96(t,3h)。
[0131]
13
c
‑
nmr(500mhz,cdcl3,ppm):153.6、150.3、143.5、142.8、129.5、112.1、109.6、93.7、64.7、34.3、32.2、31.6、29.9、20、17.3、14.8、14.4。
[0132]
实施例2
[0133]
lsy
‑3‑
o2
[0134][0135]
合成路线:
[0136][0137]
在反应瓶中加入1.13g碳酸铯(3.45mmol)、7.2mg pd2(dba)3(0.008mmol)和11mg xantphos(0.019mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽),之后将反应瓶气体置换成氮气,加入4ml无水二噁烷、0.5g 1
‑
溴
‑
3,4
‑
二氟
‑2‑
碘代苯(1.57mmol)和0.44g 3
‑
丙基环己酮(3.14mmol)。接着在80℃油浴反应24小时。待反应液冷却后,用乙醚和水进行萃取,收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a4 0.41g。
[0138]1h
‑
nmr(500mhz,cdcl3,ppm):7.13(d,1h)、6.66(d,1h)、3.50(t,1h)、2.31(m,2h)、2.21(m,2h)、1.86(m,2h)、1.79(m,1h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0139]
取0.5g化合物a4(1.51mmol)及0.054g五硫化二磷(0.38mmol)于反应瓶内,之后将反应瓶气体置换成氮气,再加入6ml无水甲苯于室温下搅拌30分钟。接着再加入0.42g六甲基二硅氧烷(2.57mmol),并且在90℃油浴反应24小时。之后待反应液冷却后,把反应液以硅
胶短管柱过滤、减压浓缩,可得产物b4 0.37g。
[0140]1h
‑
nmr(500mhz,cdcl3,ppm):7.13(d,1h)、6.66(d,1h)、2.8(t,1h)、1.7(m,2h)、1.5(m,1h)、1.40(m,2h)、1.38(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0141]
取0.5g化合物b4(1.44mmol)、0.66g碳酸铯(2.02mmol)、33mg pd2(dba)3(0.036mmol)及38.8mg dpephos(0.072mmol,(2
‑
二苯基膦苯基)醚)于反应瓶内,之后将反应瓶气体置换成氮气。接着加入4ml无水甲苯后,在100℃油浴反应20小时,待冷却后将反应液以硅藻土过滤、减压浓缩,将浓缩液以柱层析纯化,可得到产物c4 0.36g。
[0142]1h
‑
nmr(500mhz,cdcl3,ppm):7.61(d,1h)、7.00(d,1h)、2.63(m,2h)、2.50(m,2h)、1.86(m,1h)、1.71(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0143]
取0.5g化合物c(1.88mmol)溶解在13ml thf中,将反应瓶降温至
‑
78℃后加入1.3ml的含1.5m n
‑
丁基锂(1.97mmol)的己烷溶液,之后升温至0℃反应30分钟。接着在
‑
78℃下加入0.2g硼酸三甲酯(1.97mmol)后,将温度升温到室温反应1小时。之后加入1ml醋酸以及0.22ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物d4 0.45g。
[0144]1h
‑
nmr(500mhz,cdcl3,ppm):7.08(s,1h)、5.0(s,1h)、2.63(m,2h)、2.50(m,2h)、1.86(m,1h)、1.71(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0145]
取0.5g化合物d4(1.77mmol)、0.49g碳酸钾(3.54mmol)及0.33g碘乙烷(2.12mmol)于反应瓶内,加入5ml二甲基甲酰胺,在70℃油浴反应3小时,待反应液降温后以水及乙酸乙酯进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物e4 0.44g。
[0146]
制备得到的化合物e4(lsy
‑3‑
o2)进行质谱、核磁共振谱测试,测试得到的数据如下。根据测试结果,化合物e4为lsy
‑3‑
o2所示结构。
[0147]
ms(ei,m/z):222,265,310。
[0148]1h
‑
nmr(500mhz,cdcl3,ppm):7.12(s,1h)、3.98(q,2h)、2.63(m,2h)、2.60(m,2h)、1.86(m,1h)、1.71(m,2h)、1.33
‑
1.25(m,7h)、0.98(t,3h)。
[0149]
13
c
‑
nmr(500mhz,cdcl3,ppm):145.6、143.1、141.8、136、131.5、130.6、121.9、101、64.7、36.7、34.7、31.6、27.7、22.8、20.6、15.1、14.6。
[0150]
实施例3
[0151]
lsp[f,ot]
‑3‑
o2
[0152][0153]
合成路线:
[0154][0155]
在反应瓶中加入0.93g碳酸铯(2.86mmol)、5.95mg pd2(dba)3(0.065mmol)和9.23mg xantphos(0.016mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽),之后将反应瓶气体置换成氮气,加入4ml无水二噁烷、0.5g 1
‑
溴
‑3‑
氟
‑2‑
碘代
‑4‑
(三氟甲氧基)苯(1.30mmol)和0.36g 3
‑
丙基环己酮(2.6mmol)。接着在80℃油浴反应24小时。待反应液冷却后,用乙醚和水进行萃取,收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a5 0.40g。
[0156]1h
‑
nmr(500mhz,cdcl3,ppm):7.04(d,1h)、6.46(d,1h)、3.50(t,1h)、2.31(m,2h)、2.21(m,2h)、1.88(m,2h)、1.79(m,1h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0157]
取0.5g化合物a5(1.26mmol)及0.045g五硫化二磷(0.31mmol)于反应瓶内,之后将反应瓶气体置换成氮气,再加入5ml无水甲苯于室温下搅拌30分钟。接着再加入0.35g六甲基二硅氧烷(2.14mmol),并且在90℃油浴反应24小时。之后待反应液冷却后,把反应液以硅胶短管柱过滤、减压浓缩,可得产物b5 0.36g。
[0158]1h
‑
nmr(500mhz,cdcl3,ppm):7.04(d,1h)、6.46(d,1h)、2.8(t,1h)、1.7(m,2h)、1.5(m,1h)、1.4(m,2h)、1.38(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0159]
取0.5g化合物b5(1.21mmol)、0.55g碳酸铯(1.69mmol)、27.7mg pd2(dba)3(0.03mmol)及32.6mg dpephos(0.061mmol,(2
‑
二苯基膦苯基)醚)于反应瓶内,之后将反应瓶气体置换成氮气。接着加入4ml无水甲苯后,在100℃油浴反应20小时,待冷却后将反应液以硅藻土过滤、减压浓缩,将浓缩液以柱层析纯化,可得到产物c5 0.38g。
[0160]1h
‑
nmr(500mhz,cdcl3,ppm):7.52(d,1h)、6.8(d,1h)、2.63(m,2h)、2.5(m,2h)、1.86(m,1h)、1.71(m,2h)、1.35
‑
1.24(m,4h)、0.99(t,3h)。
[0161]
取0.5g化合物c5(1.5mmol)溶解在10ml thf中,将反应瓶降温至
‑
78℃后加入1.05ml的含1.5m n
‑
丁基锂(1.58mmol)的己烷溶液,之后升温至0℃反应30分钟。接着在
‑
78℃下加入0.16g硼酸三甲酯(1.58mmol)后,将温度回到室温反应1小时。之后加入1ml醋酸以及0.18ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物d5 0.44g。
[0162]1h
‑
nmr(500mhz,cdcl3,ppm):6.99(s,1h)、5.0(s,1h)、2.63(m,2h)、2.60(m,2h)、1.86(m,1h)、1.71(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0163]
取0.5g化合物d5(1.44mmol)、0.4g碳酸钾(2.88mmol)及0.27g碘乙烷(1.73mmol)于反应瓶内,加入5ml二甲基甲酰胺,在70℃油浴反应3小时,待反应液降温后以水及乙酸乙
酯进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物e5 0.43g。制备得到的化合物e(lsp(fot)
‑3‑
o2)进行质谱、核磁共振谱测试,测试得到的数据如下。根据测试结果,化合物e5为lsp(fot)
‑3‑
o2所示结构。
[0164]
ms(ei.m/z):288,333,376。
[0165]1h
‑
nmr(500mhz,cdcl3,ppm):7.03(s,1h)、3.98(q,2h)、2.63(m,2h)、2.60(m,2h)、1.86(m,1h)、1.71(m,2h)、1.35
‑
1.26(m,7h)、0.97(t,3h)。
[0166]
13
c
‑
nmr(500mhz,cdcl3):147.6、143.6、141.8、132.7、131.8、130.1、122.1、121.3、101.4、66.2、36.7、34.7、31.6、27.7、22.8、20.6、15.2、14.6。
[0167]
实施例4
[0168]
lsp[to,ot]
‑3‑
o2
[0169][0170]
合成路线:
[0171][0172]
在反应瓶中加入0.80g碳酸铯(2.44mmol)、5.08mg pd2(dba)3(0.0056mmol)和7.7mg xantphos(0.133mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽),之后将反应瓶气体置换成氮气,加入3ml无水二噁烷、0.5g 1
‑
溴
‑2‑
碘
‑
3,4
‑
双(三氟甲氧基)苯(1.11mmol)和0.31g 3
‑
丙基环己酮(2.22mmol)。接着在80℃油浴反应24小时。待反应液冷却后,用乙醚和水进行萃取,收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a6 0.40g。
[0173]1h
‑
nmr(500mhz,cdcl3,ppm):6.83(d,1h)、6.37(d,1h)、3.50(t,1h)、2.36(m,2h)、2.21(m,2h)、1.89(m,2h)、1.77(m,1h)、1.36
‑
1.24(m,4h)、0.97(t,3h)。
[0174]
取0.5g化合物a6(1.08mmol)及0.038g五硫化二磷(0.27mmol)于反应瓶内,之后将反应瓶气体置换成氮气,再加入5ml无水甲苯于室温下搅拌30分钟。接着再加入0.3g六甲基二硅氧烷(1.84mmol),并且在90℃油浴反应24小时。之后待反应液冷却后,把反应液以硅胶短管柱过滤、减压浓缩,可得产物b6 0.34g。
[0175]1h
‑
nmr(500mhz,cdcl3,ppm):6.85(d,1h)、6.38(d,1h)、2.8(t,1h)、1.7(m,2h)、1.5(m,1h)、1.4(m,2h)、1.38(m,2h)、1.33
‑
1.25(m,4h)、0.95(t,3h)。
[0176]
取0.5g化合物0.5g化合物b(1.04mmol)、0.48g碳酸铯(1.46mmol)、24mg pd2(dba)3(0.026mmol)及28mg dpephos(0.052mmol,(2
‑
二苯基膦苯基)醚)于反应瓶内,之后将反应瓶气体置换成氮气。接着加入3ml无水甲苯后,在100℃油浴反应20小时,待冷却后将反应液以硅藻土过滤、减压浓缩,将浓缩液以柱层析纯化,可得到产物c6 0.4g。
[0177]1h
‑
nmr(500mhz,cdcl3,ppm):7.31(d,1h)、6.71(d,1h)、2.63(m,2h)、2.50(m,2h)、1.86(m,1)、1.7(m,2h)、1.34
‑
1.23(m,4h)、0.96(t,3h)。
[0178]
取0.5g化合物c6(1.2 6mmol)溶解在10ml thf中,将反应瓶降温至
‑
78℃后加入1.05ml的含1.5m n
‑
丁基锂(1.32mmol)的丁烷,之后升温至0℃反应30分钟。接着在
‑
78℃下加入0.14g硼酸三甲酯(1.32mmol)后,将温度回到室温反应1小时。之后加入1ml醋酸以及0.15ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物d6 0.45g。
[0179]1h
‑
nmr(500mhz,cdcl3,ppm):6.78(s,1h)、5.0(s,1h)、2.63(m,2h)、2.50(m,2h)、1.86(m,1h)、1.7(m,2h)、1.33
‑
1.25(m,4h)、0.96(t,3h)。
[0180]
取0.5g化合物d6(1.21mmol)、0.33g碳酸钾(2.42mmol)及0.23g碘乙烷(1.45mmol)于反应瓶内,加入5ml二甲基甲酰胺,在70℃油浴反应3小时,待反应液降温后以水及乙酸乙酯进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物e6 0.38g。制备得到的化合物进行质谱、核磁共振谱测试,测试得到的数据如下。根据测试结果,化合物e6为lsp(toot)
‑3‑
o2所示结构。
[0181]
ms(ei,m/z):356,399,442。
[0182]1h
‑
nmr(500mhz,cdcl3,ppm):6.82(s,1h)、3.98(q,2h)、2.63(m,2h)、2.60(m,2h)、1.86(m,1h)、1.71(m,2h)、1.32
‑
1.23(m,7h)、0.98(t,3h)。
[0183]
13
c
‑
nmr(500mhz,cdcl3):147、142.5、140.8、132.1、131.1、129.1、122.5、122.4、119.9、97.1、65、36.7、34.7、31.6、27.2、22.7、20.6、14.6、13.9。
[0184]
实施例5
[0185]
lsp[t,f]
‑3‑
o2
[0186][0187]
合成路线:
[0188][0189]
取1.0g 3
‑
丙基环己
‑1‑
酮(7.14mmol)、1.25g 1
‑
溴
‑4‑
氟
‑2‑
碘
‑3‑
(三氟甲基)苯(3.39mmol)、2.33g碳酸铯(7.14mmol)、73mg pd2(dba)3(0.08mmol)以及14.8mg xantphos(0.16mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽)于反应瓶中,并且将气体置换成氮气。接着加入15ml无水二噁烷进行搅拌,然后加热至90℃、反应24小时。等待温度回到室温后,将反应液以乙酸乙酯以及水进行萃取、收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a7 0.93g。
[0190]1h
‑
nmr(500mhz,cdcl3,ppm):7.52(d,1h)、7.02(d,1h)、3.5(t,1h)、2.31~2.21(m,2h)、2.06~1.96(m,2h)、1.72(m,2h)、1.48(q,1h)、1.32(q,2h)、1.18(q,2h)、0.91(t,3h)。
[0191]
取0.5g化合物a7(1.31mmol)及73mg五硫化二磷(0.33mmol)于反应瓶中,之后将气体置换成氮气,加入8ml无水甲苯,室温下搅拌30分钟。接着再加入0.36g六甲基二硅氧烷(2.23mmol),移至90℃油浴下搅拌24小时。待反应液冷却后,将反应液以硅胶短管柱过滤,后续把溶液减压浓缩,可得到产物b7 0.32g。
[0192]1h
‑
nmr(500mhz,cdcl3,ppm):7.55(d,1h)、7.08(d,1h)、2.75(t,1h)、1.73(q,1h)、1.51(q,1h)、1.35~1.18(m,7h)、1.02(q,2h)、0.91(t,3h)。
[0193]
取0.5g化合物b7(1.26mmol)、0.61g碳酸铯(1.89mmol)、28.8mg pd2(dba)3(0.031mmol)及33.9mg dpephos(0.063mmol,(2
‑
二苯基膦苯基)醚)于反应瓶,然后把气体置换成氮气、加入5ml无水甲苯,在100℃油浴下反应20小时。待反应液冷却后,将反应液以硅藻土短管柱过滤、减压浓缩,接着把浓缩液以柱层析纯化,可得到产物c7 0.3g。
[0194]1h
‑
nmr(500mhz,cdcl3,ppm):7.98(d,1h)、7.15(d,1h)、2.88~2.8(m,3h)、2.58(d,1h)、1.68~1.56(m,2h)、1.35~1.18(m,5h)、0.88(t,3h)。
[0195]
取0.5g化合物c7(1.58mmol)溶解在10ml thf中,将反应瓶降温至
‑
78℃后加入1.11ml含1.5m n
‑
丁基锂(1.66mmol)的丁烷,之后由
‑
78℃升温至0℃反应30分钟。接着在
‑
78℃下加入0.17g硼酸三甲酯(1.32mmol)后,将温度升至室温反应1小时。之后加入1ml醋酸以及0.2ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物d70.39g。
[0196]1h
‑
nmr(500mhz,cdcl3,ppm):8.9(s,1h)、7.20(s,1h)、2.8~2.81(m,3h)、2.62(d,
1h)、1.70~1.58(m,2h)、1.33~1.20(m,5h)、0.89(t,3h)。
[0197]
取0.5g化合物d7(1.50mmol)、0.196g溴乙烷(1.80mmol)及8ml thf于反应瓶,然后再滴入0.23g三乙胺(2.25mmol),最后加热回流反应3小时。待反应液降温后以乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。接着将浓缩物进行柱层析,可得到产物e7 0.47g。
[0198]
制备得到的化合物e7进行质谱、核磁共振谱测试,测试得到的数据如下。根据测试结果,化合物e7为lsp(tf)
‑3‑
o2所示结构。
[0199]
ms(ei,m/z):272,315,360。
[0200]1h
‑
nmr(500mhz,cdcl3,ppm):7.41(s,1h)、4.06(q,2h)、2.8~2.82(m,3h)、2.62(d,1h)、1.69~1.58(m,2h)、1.33~1.20(m,8h)、0.88(t,3h)。
[0201]
13
c
‑
nmr(500mhz,cdcl3,ppm):142、141.7、140.4、134.1、131.7、115.4、113.9、108.6、64.6、38.8、34.6、32.3、27.5、22.6、20.5、14.8、14.2。
[0202]
实施例6
[0203]
lsp[t,t]
‑3‑
o2
[0204][0205]
合成路线:
[0206][0207]
取1.0g 3
‑
丙基环己
‑1‑
酮(7.14mmol)、1.42g 1
‑
溴
‑2‑
碘
‑
3,4
‑
双(三氟甲基)苯(3.39mmol)、2.33g碳酸铯(7.14mmol)、73mg pd2(dba)3(0.08mmol)以及14.8mg xantphos(0.16mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽)于反应瓶中,并且将气体置换成氮气。接着加入15ml无水二噁烷进行搅拌,然后加热至90℃、反应24小时。等待温度回到室温后,将反应液以乙酸乙酯以及水进行萃取、收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a8 1.08g。
[0208]1h
‑
nmr(500mhz,cdcl3,ppm):7.51(d,1h)、7.30(d,1h)、3.52(t,1h)、2.32~2.21
(m,2h)、2.05~1.96(m,2h)、1.73(m,2h)、1.51(q,1h)、1.32(q,2h)、1.21(q,2h)、0.90(t,3h)。
[0209]
取0.5g化合物a8(1.16mmol)及64.4mg五硫化二磷(0.29mmol)于反应瓶内,之后将气体置换成氮气,加入8ml无水甲苯、室温下搅拌30分钟。接着再加入0.32g六甲基二硅氧烷(1.97mmol),移至90℃油浴下搅拌24小时。待反应液冷却后,将反应液以硅胶短管柱过滤,接着把溶液减压浓缩,可得到产物b8 0.36g。
[0210]1h
‑
nmr(500mhz,cdcl3,ppm):7.49(d,1h)、7.29(d,1h)、2.72(t,1h)、1.71(q,1h)、1.55(q,1h)、1.32~1.18(m,7h)、1.04(m,2h)、0.89(t,3h)。
[0211]
取0.5g化合物b8(1.12mmol)、0.55g碳酸铯(1.68mmol)、25.6mg pd2(dba)3(0.028mmol)及30.2mg dpephos(0.056mmol,(2
‑
二苯基膦苯基)醚)于反应瓶,然后把气体置换成氮气、加入5ml无水甲苯,在100℃油浴下反应20小时。待反应液冷却后,将反应液以硅藻土短管柱过滤、减压浓缩,接着把浓缩液以柱层析纯化,可得到产物c8 0.24g。
[0212]1h
‑
nmr(500mhz,cdcl3,ppm):7.76(d,1h)、7.51(d,1h)、2.90~2.79(m,3h)、2.65(d,1h)、1.68~1.56(m,2h)、1.33~1.20(m,5h)、0.91(t,3h)。
[0213]
取0.5g化合物c8(1.36mmol)溶解在10ml thf中,将反应瓶降温至
‑
78℃后加入0.95ml含1.5m n
‑
丁基锂(1.43mmol)的己烷,之后由
‑
78℃升温至0℃反应30分钟。接着在
‑
78℃下加入0.16g硼酸三甲酯(1.5mmol)后,将温度回到室温反应1小时。之后加入1ml醋酸以及0.15ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物d80.43g。
[0214]1h
‑
nmr(500mhz,cdcl3,ppm):9.69(br,1h)、7.21(s,1h)、2.91~2.80(m,3h)、2.63(d,1h)、1.68~1.58(m,2h)、1.35~1.19(m,5h)、0.91(t,3h)。
[0215]
取0.5g化合物d8(1.31mmol)、0.17g溴乙烷(1.57mmol)及8ml thf于反应瓶,然后再滴入0.20g三乙胺(1.97mmol),最后加热回流反应3小时。待反应液降温后以乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。接着将浓缩物进行柱层析,可得到产物e8 0.43g。
[0216]
制备得到的化合物e8进行质谱、核磁共振谱测试,测试得到的数据如下。根据测试结果,化合物e8为lsp(tt)
‑3‑
o2所示结构。
[0217]
ms(ei,m/z):322,381,410。
[0218]1h
‑
nmr(500mhz,cdcl3,ppm):7.35(s,1h)、4.8(q,2h)、2.8~2.83(m,3h)、2.61(d,1h)、1.69~1.57(m,2h)、1.33~1.20(m,8h)、0.89(t,3h)。
[0219]
13
c
‑
nmr(500mhz,cdcl3,ppm):151.8、143.8、131.5、130.4、123.7、119.7、116.1、109.5、107.3、64.9、38.8、35.8、31.5、27.5、21.6、20.5、14.8、13.9。
[0220]
实施例7
[0221]
cloy
‑3‑
o2
[0222][0223]
合成路线:
[0224][0225]
取1.0g 4
′‑
丙基
‑
[1,1
′‑
二(环己烷)]
‑3‑
酮(4.5mmol)、0.68g 1
‑
溴
‑4‑
氟
‑2‑
碘
‑3‑
(三氟甲基)苯(2.14mmol)、1.46g碳酸铯(4.49mmol)、49mg pd2(dba)3(0.08mmol)以及62mg xantphos(0.11mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽)于反应瓶中,并且将气体置换成氮气。接着加入15ml无水二噁烷进行搅拌,然后加热至90℃、反应24小时。等待温度回到室温后,将反应液以乙酸乙酯以及水进行萃取、收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a9 0.64g。
[0226]1h
‑
nmr(500mhz,cdcl3,ppm):7.33(d,1h)、7.12(d,1h)、3.55(t,1h)、2.3~2.22(m,2h)、2.08~1.98(m,2h)、1.78~1.2(m,17h)、0.92(t,3h)。
[0227]
取0.5g化合物a9(1.21mmol)、0.59g碳酸铯(1.82mmol)、22.9mg pd2(dba)3(0.028mmol)及32.6mg dpephos(0.061mmol,(2
‑
二苯基膦苯基)醚)于反应瓶,然后把气体置换成氮气、加入5ml无水甲苯,在100℃油浴下反应20小时。待反应液冷却后,将反应液以硅藻土短管柱过滤、减压浓缩,接着把浓缩液以柱层析纯化,可得到产物b9 0.34g。
[0228]1h
‑
nmr(500mhz,cdcl3,ppm):7.38(d,1h)、6.82(d,1h)、2.87~2.75(q,2h)、2.46(d,1h)、2.30(d,1h)、1.66~1.21(m,17h)、0.91(t,3h)。
[0229]
0.5g化合物b9(1.50mmol)溶解在10ml thf中,将反应瓶降温至
‑
78℃后加入1.05ml 1.5m n
‑
丁基锂in己烷(1.58mmol),之后由
‑
78℃升温至0℃反应30分钟。接着在
‑
78℃下加入0.17g硼酸三甲酯(1.65mmol)后,将温度回到室温反应1小时。之后加入1ml醋酸以及0.17ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物c9 0.46g。
[0230]1h
‑
nmr(500mhz,cdcl3,ppm):9.63(s,1h)、7.15(d,1h)、2.87~2.74(m,2h)、2.48(d,1h)、2.23(d,1h)、1.72~1.3(m,17h)、0.91(t,3h)。
[0231]
取0.5g化合物d9(1.44mmol)、0.19g溴乙烷(1.73mmol)及8ml thf于反应瓶,然后再滴入0.22g三乙胺(2.16mmol),最后加热回流反应3小时。待反应液降温后以乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。接着将浓缩物进行柱层析,可得到产物d9 0.45g。
[0232]
制备得到的化合物d9进行质谱、核磁共振谱测试,测试得到的数据如下。根据测试
结果,化合物为cloy
‑3‑
o2所示结构。
[0233]
ms(ei,m/z):206,251,376。
[0234]1h
‑
nmr(500mhz,cdcl3,ppm):7.33(s,1h)、4.13(q,2h)、2.90~2.78(q,2h)、2.47(d,1h)、2.23(d,1h)、1.71~1.31(m,20h)、0.93(t,3h)。
[0235]
13
c
‑
nmr(500mhz,cdcl3,ppm):155.4、150.5、143.5、142.8、129.5、112.1、109.6、93.6、64.6、40.1、37.3、35.8、30.3、29.3、26.8、20.5、17.1、15.1、14.4。
[0236]
实施例8
[0237]
clsy
‑3‑
o2
[0238][0239]
合成路线:
[0240][0241]
取1.0g 4
′‑
丙基
‑
[1,1
′‑
二(环己烷)]
‑3‑
酮(2.5mmol)、0.68g 1
‑
溴
‑4‑
氟
‑2‑
碘
‑3‑
(三氟甲基)苯(2.14mmol)、1.46g碳酸铯(4.49mmol)、49mg pd2(dba)3(0.08mmol)以及62mg xantphos(0.11mmol,4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽)于反应瓶内,并且将气体置换成氮气。接着加入15ml无水二噁烷进行搅拌,然后加热至90℃、反应24小时。等待温度回到室温后,将反应液以乙酸乙酯以及水进行萃取、收集有机层,之后将有机层以无水mgso4除水、减压浓缩。将浓缩物进行柱层析,可得到产物a10 0.64g。
[0242]1h
‑
nmr(500mhz,cdcl3,ppm):7.33(d,1h)、7.12(d,1h)、3.55(t,1h)、2.3~2.22(m,2h)、2.08~1.98(m,2h)、1.78~1.2(m,17h)、0.92(t,3h)。
[0243]
取0.5g化合物a10(1.21mmol)及67.2mg五硫化二磷(0.30mmol)于反应瓶中,之后将气体置换成氮气,加入8ml无水甲苯,室温下搅拌30分钟。接着再加入0.33g六甲基二硅氧烷(2.06mmol),移至90℃油浴下搅拌24小时。待反应液冷却后,将反应液以硅胶短管柱过
滤,接着把溶液减压浓缩,可得到产物b10 0.52g。
[0244]1h
‑
nmr(500mhz,cdcl3,ppm):7.33(d,1h)、7.11(d,1h)、2.76(t,1h))、1.82~1.05(m,21h)、0.91(t,3h)。
[0245]
取0.5g化合物b10(1.16mmol)、0.57g碳酸铯(1.74mmol)、29mg pd2(dba)3(0.029mmol)及31.2mg dpephos(0.058mmol,(2
‑
二苯基膦苯基)醚)于反应瓶,然后把气体置换成氮气、加入5ml无水甲苯,在100℃油浴下反应20小时。待反应液冷却后,将反应液以硅藻土短管柱过滤、减压浓缩,接着把浓缩液以柱层析纯化,可得到产物c10 0.27g。
[0246]1h
‑
nmr(500mhz,cdcl3,ppm):7.82(d,1h)、7.18(d,1h)、2.92~2.81(m,3h)、2.58(d,1h)、1.66~1.17(m,17h)、0.91(t,3h)。
[0247]
取0.5g化合物c10(1.43mmol)溶解在10ml thf中,将反应瓶降温至
‑
78℃后加入1.0ml含1.5m n
‑
丁基锂(1.50mmol)的己烷,之后由
‑
78℃升温至0℃反应30分钟。接着在
‑
78℃下加入0.16g硼酸三甲酯(1.57mmol)后,将温度回到室温反应1小时。之后加入1ml醋酸以及0.18ml 30%h2o2,接着继续搅拌至隔日。反应液用乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。将得到的浓缩液进行柱层析,可得到产物d100.4g。
[0248]1h
‑
nmr(500mhz,cdcl3,ppm):9.23(s,1h)、7.15(s,1h)、2.92~2.83(m,3h)、2.60(d,1h)、1.66~1.17(m,17h)、0.92(t,3h)。
[0249]
取0.5g化合物d10(1.37mmol)、0.18g溴乙烷(1.65mmol)及8ml thf于反应瓶,然后再滴入0.21g三乙胺(2.06mmol),最后加热回流反应3小时。待反应液降温后以乙酸乙酯以及水进行萃取,之后将有机层以无水mgso4除水、减压浓缩。接着将浓缩物进行柱层析,可得到产物e10 0.43g。
[0250]
制备得到的化合物e10进行质谱、核磁共振谱测试,测试得到的数据如下。根据测试结果,化合物为clsy
‑3‑
o2所示结构。
[0251]
ms(ei,m/z):222,349,392。
[0252]1h
‑
nmr(500mhz,cdcl3,ppm):7.18(s,1h)、4.13(q,2h)、2.92~2.81(m,3h)、2.63(d,1h)、1.66~1.17(m,20h)、0.90(t,3h)。
[0253]
13
c
‑
nmr(500mhz,cdcl3,ppm):145.5、143.3、141.8、135.4、131.5、129.6、121.7、100.9、64.6、42.6、39.7、37.1、30.9、29.3、26.8、25.3、22.6、20.5、15.5、13.8。
[0254]
对于下述的表1所示的前述实施例及对比例的各化合物单体,将其与下述的母体液晶以重量百分比计按照10%化合物单体与90%母体液晶的比例混合相容后,在下述的条件下测量t
ni
、δn、δε、k
11
、k
33
、g1,然后采用外插法计算得到单体的t
ni
、δn、δε、k
11
、k
33
、g1,得到的单体的各物理性能结果示于后述的表2中。
[0255]
前述的母体液晶中各组分及其质量百分含量如下:
[0256]
ccg
‑2‑
f10%ccp
‑
v
‑
120%ccp
‑
v2
‑
120%cpu
‑3‑
f20%cp
‑3‑
o215%cp
‑3‑
o115%
[0257]
根据这些测试结果计算得到的响应指标值g1/(k
11
*
△
n*
△
n)、g1/(k
33
*
△
n*
△
n)
示于后述的表3中,在va(vertical alignment,垂直取向)或者ps
‑
va(polymer stabilized vertical alignment,聚合物稳定垂直取向)模式下,液晶介质的响应时间与指标g1/(k
33
*
△
n*
△
n)相关,而在ffs(fringe field switching,边缘场开关)、ips(in
‑
plane switching,平面转换)、ps
‑
ffs(polymer stabilized fringe field switching,聚合物稳定边缘场开关)、ps
‑
ips(polymer stabilized in
‑
plane switching,聚合物稳定平面转换)等模式下,液晶介质的响应时间与响应指标值g1/(k
11
*
△
n*
△
n)相关。前述的响应指标值越小,表明响应时间越快。
[0258]
t
ni
代表液晶单体由向列相相变至澄清相之温度,其温度通过mp
‑
90设备测量;
[0259]
δn表示光学各向异性,δn=n
e
‑
n
o
,其中,n
o
为寻常光的折射率,n
e
为非寻常光的折射率,测试条件:589nm、25
±
0.2℃。
[0260]
δε表示介电各向异性,δε=ε
||
‑
ε
⊥
,其中,ε
||
为平行于分子轴的介电常数,ε
⊥
为垂直于分子轴的介电常数,测试条件:25℃、instec:alct
‑
ir1、18微米垂直盒;
[0261]
k
11
为扭曲弹性常数,k
33
为展曲弹性常数,测试条件为:25℃、instec:alct
‑
ir1、18微米垂直盒。
[0262]
gamma1(mpa.s)为旋转粘滞系数,简写为“g1”,测试条件为:25℃、instec:alct
‑
ir1、18微米垂直盒。
[0263]
表1:实施例及对比例的各化合物
[0264][0265][0266]
表2:实施例及对比例的各化合物的物理性能结果
[0267][0268]
表3:实施例及对比例的各化合物的响应指标值
[0269][0270][0271]
通过表3中实施例1~8以及对比例1~2的响应指标指的对比可以看出,实施例1~
8的液晶化合物的响应指标值g1/(k
11
*
△
n*
△
n)、g1/(k
33
*
△
n*
△
n)相对于对比例降低。
[0272]
本发明可用其他的不违背本发明的精神或主要特征的具体形式来概述。因此,无论从哪一点来看,本发明的上述实施方案都只能认为是对本发明的说明而不能限制本发明,权利要求书指出了本发明的范围,而上述的说明并未指出本发明的范围,因此,在与本发明的权利要求书相当的含义和范围内的任何改变,都应认为是包括在本发明的权利要求书的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。