1.本发明属于医药化工领域,具体涉及一种月桂醇的精制工艺及以精制品为原料制备聚桂醇的工艺。
背景技术:
2.聚桂醇(lauromacrogol),又名乙氧硬化醇、聚多卡醇,分子式c
12
h
25
(och2ch2)
n
oh(n=9),是由月桂醇(脂肪醇)和多个环氧乙烷聚合得到的聚合物产品,是目前欧美国家临床应用最广泛的硬化药物,主要应用于静脉曲张、静脉畸形、血管瘤、内痔、囊肿性疾病等的硬化治疗。
3.聚桂醇的安全性已被国际医疗界公认,是德国迄今唯一被批准用于硬化治疗的药物,并已经在其他欧洲国家应用,美国食品药品管理局于2010年3月批准聚桂醇上市,我国于2008年开始临床应用聚桂醇(陕西天宇制药公司2008年上市生产,剂型为注射液)。
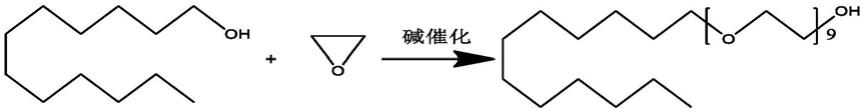
4.聚桂醇的结构式如下:
[0005][0006]
聚桂醇主要通过月桂醇与环氧乙烷在碱催化条件下发生聚合反应制备得到,反应过程如下:
[0007][0008]
发明人在研究过程中发现,聚桂醇产品的质量和稳定性受到起始物料纯度、反应催化剂、反应压力、后处理工艺等的影响,其中,如何提高聚桂醇原料药产品在长期存放过程中的稳定性以及减少聚桂醇中的杂质含量,是保证聚桂醇原料药安全性和有效性的关键。
[0009]
按现有工艺制备的聚桂醇产品的稳定性较差,在室温下就会发生降解,空气中的氧气会加速这一降解过程,导致生成大量未知杂质。罗氏制药在售的聚桂醇原料的存储条件为2
‑
8℃,氩气或氮气保护,避光储存。发明人同时以采购的usp聚桂醇、中检院聚桂醇(标注为拆封后迅速用完)为对照品,进行稳定性试验,结果发现中检院聚桂醇对照品室温下降解明显,usp聚桂醇对照品虽然稳定性略好,但是在10天内也明显降解出了较大杂质。由于目前市售聚桂醇产品不稳定,需要通过低温、隔绝空气的方式进行存储,存储条件苛刻,对储存设备的要求高,储存成本高,且样品一旦开封,需要迅速使用完毕,即便如此,也无法完全避免样品的降解,不利于用药的安全。此外,文献中的聚桂醇制备工艺中均包含月桂醇减压脱水步骤,在高湿度的南方地区,水分很难降至文献要求之下,造成大量能源的消耗和成本的提升。
[0010]
按照现有工艺制备的聚桂醇产品中,主要包含三类杂质,分别为反应剩余的起始物料月桂醇、月桂醇引入的杂质(如:正十醇、正十四醇、未知杂质)与环氧乙烷反应生成的杂质以及过程中引入的游离聚乙二醇、二氧六环和二甘醇等化合物。由于产品为粘状液体(24℃),且在大部分的有机溶剂中溶解性较好,导致杂质很难进行去除。
[0011]
通过实验研究发现:陕西天宇制药有限公司的聚桂醇制备方法(cn1762948a)得到的产品不能完全符合欧洲药典的质量标准;通过南京正大天晴制药有限公司的聚桂醇精制方法(cn103922901a)得到的产品可以有效去除起始物料月桂醇,但不能有效的去除其他杂质,且需要进行高温(130℃~160℃)和高真空(
‑
0.1mpa)蒸馏,工业上不易实现;通过北京万诚伟业医药科技有限公司月桂醇纯化方法与聚桂醇制备方法(cn104649863a)得到的产品虽然可以达到要求,但是其采用减压蒸馏的方式对月桂醇进行纯化,月桂醇纯化需要多次重复精制,而且反应条件为高温(150℃~160℃)和高真空(
‑
0.1mpa),因此对设备要求很高,多次蒸馏能耗很大,很难实现工业化。按照上述专利及专利cn110015950a中的聚桂醇制备工艺所制备出的聚桂醇产品,在室温及氧气存在下不稳定,很快降解出大量未知杂质,导致产品在放置过程中的质量无法保障。
[0012]
鉴于聚桂醇原料的临床药用价值及良好的市场前景,解决目前聚桂醇原料药稳定性较差的问题,降低储存难度和存储成本,提高聚桂醇原料药的稳定性和质量,具有非常重要的意义。
技术实现要素:
[0013]
本发明的目的在于提供一种操作简单、反应条件温和、适用于工业规模化生产的聚桂醇的生产工艺,以解决目前聚桂醇原料药稳定性较差的问题,降低储存难度和存储成本,提高聚桂醇原料药的稳定性和质量。
[0014]
第一方面,本发明提供一种月桂醇的精制工艺,创造性的采用有机溶剂重结晶的方式对月桂醇原料进行精制。由于月桂醇室温下为粘稠液体,导致通常技术人员很难想到通过重结晶析出固体并在室温下进行抽滤的方式,对月桂醇粘稠液体进行精制。本发明通过选用合适的溶剂,优选进一步严格控制析晶温度,从而保证月桂醇能够在溶剂中以固体的形式析出,通过筛选溶剂的配比,保证了所析出月桂醇产品的收率和质量,所述月桂醇的精制工艺中,所述有机溶剂为乙醇、甲醇、异丙醇、四氢呋喃、丙酮、乙腈、甲苯、二甲苯、正己烷、环己烷、异辛烷、正戊烷和石油醚中的至少一种,优选为选自于四氢呋喃、丙酮、乙腈、甲苯、环己烷和石油醚中的至少一种;或者,优选为乙醇、甲醇、异丙醇、四氢呋喃、丙酮、乙腈、甲苯、二甲苯、正己烷、环己烷、异辛烷、正戊烷和石油醚中的至少两种,更优选为四氢呋喃、丙酮、乙腈、甲苯、环己烷和石油醚中的至少两种。在所有情况下,有机溶剂包含两种时,两种之间的质量比可以为1:0.25~4.0,优选1:0.5~2,优选1:0.8~1.2,例如质量比1:0.25~4.0,优选1:0.5~2.0,优选1:0.8~1.2的四氢呋喃和乙腈、或者四氢呋喃和甲苯。在优选的实施方案中,本发明通过选择合理的溶剂配比,达到月桂醇在溶剂中低温(例如至
‑
5℃~10℃,进一步
‑
3~8℃,更优选0
‑
5℃)下能够以固体的形式析出、且析出的月桂醇固体能够满足过滤过程中不会在所选用溶剂中溶解的要求,从而保证整个精制过程的顺利完成,固体析出后优选进一步经历抽滤和减压蒸馏而获得产品。
[0015]
本技术中所述的月桂醇原料可通过从市场上购买,例如,本技术中的月桂醇可采
购自上海嘉辰化工有限公司、含量大于98%的月桂醇产品,通常含有正癸醇0.10
‑
0.50%,进一步0.20
‑
0.30%,正十四醇0.1
‑
0.50%,进一步0.20
‑
0.30%和未知杂质0.01
‑
0.50%。也可使用按照现有技术合成或提取的月桂醇原料。
[0016]
优选地,上述月桂醇的精制工艺,包括下述步骤:
[0017]
1)将月桂醇原料、有机溶剂加入至反应容器中,升温至20℃以上,例如25~45℃,优选25℃~35℃,搅拌至溶解,得到反应液;
[0018]
2)将所述反应液降温至
‑
5℃~10℃,进一步
‑
3~8℃,更优选0
‑
5℃,搅拌析晶例如10~60分钟,进一步20
‑
40分钟,优选30分钟,抽滤,收集滤饼,得到中间体;
[0019]
3)将所述中间体加入至反应容器中,缓慢升温至20~80℃,优选30
‑
60℃,更优选35
‑
45℃,例如约40℃,搅拌至至融化优选全融,减压蒸馏,蒸出体系中残留的有机溶剂,收集蒸馏产物,得到剩余物,即为目标产品月桂醇精制品。通过重结晶步骤对月桂醇原料进行处理,得到的月桂醇精制品相对于精制前,纯度明显改善,特别是杂质正十醇和正十四醇的含量明显降低,已知杂质和未知杂质均降低至0.1%以下,满足目前对于医药中间体的要求。
[0020]
优选地,上述月桂醇的精制工艺中:
[0021]
步骤1)中,月桂醇原料与有机溶剂的重量比可以为1:1~4,优选1:1~3或1:1~2.5,优选地,所述有机溶剂为四氢呋喃和乙腈的混合溶剂,四氢呋喃和乙腈的重量比可以为(0.5~2):(0.5~2),进一步(0.5~1):(0.5~2),优选地,月桂醇原料、四氢呋喃和乙腈三者重量比为1:(0.5~2):(0.5~2),优选为1:(0.5~1):(0.5~2),例如1:0.5:0.5、1:0.5:1、1:0.5:2、1:1:2,更优选为1:1:1;和/或,
[0022]
步骤2)中,降温至
‑
5℃~10℃,优选
‑
3℃~2℃;和/或,
[0023]
步骤3)中,所述减压蒸馏的具体操作为:逐渐升温至80℃~100℃,控制真空度≤
‑
0.1mpa,优选为
‑
0.1mpa至0.05mpa或
‑
0.1mpa,减压蒸馏2h~6h,优选为4h~5h。
[0024]
第二方面,本发明提供一种月桂醇精制品,其由上述月桂醇的精制工艺制得;优选地,所述月桂醇精制品中所含单杂≤0.1wt%,优选≤0.05wt%,优选正十醇≤0.1wt%,进一步正十醇≤0.05wt%、正十四醇≤0.1wt%,进一步正十四醇≤0.05wt%和/或未知杂质≤0.1wt%,进一步未知杂质≤0.05wt%。
[0025]
第三方面,本发明提供一种聚桂醇的制备工艺,其以上述精制工艺制得的月桂醇精制品为原料进行制备。
[0026]
优选地,上述聚桂醇的制备工艺,包括以下步骤:上述精制工艺制得的月桂醇精制品以碱优选氢氧化钾为催化剂进行聚合反应,氮气增压后通入环氧乙烷,升温、持续通入环氧乙烷,停止加热、氮气置换、排空,重新通入氮气增压,反应完毕降温,溶于乙腈、趁热过滤,离心分离。
[0027]
优选地,上述聚桂醇的制备工艺,包括以下步骤:
[0028]
(1)向高压反应釜中,加入上述精制工艺制得的月桂醇精制品和0.003
‑
0.05摩尔当量的碱优选氢氧化钾,锁紧釜盖,搅拌5~20min优选10min,转速为12
‑
15rpm,通入氮气至0.1~0.5mpa优选0.2mpa,试漏,升温至80
‑
100℃,氮气置换1
‑
5次优选3次;确保体系中空气置换充分,采用氮气置换3次的操作方式,使体系中氮气置换更充分,有效的减少体系中氧气的存在,避免了产品氧化值超标;
[0029]
(2)氮气增压至0.05
‑
0.4mpa优选0.15mpa,缓慢通入环氧乙烷增压至0.1
‑
0.5mpa优选0.20
‑
0.25mpa;通过氮气增压,一方面使体系可以保持反应所需压力,同时体系中的氮气,在缓慢通入环氧乙烷时,有效降低了体系内环氧乙烷的浓度,有利于反应的控制,保证聚合的质量;
[0030]
(3)升温至140~150℃,压力保持在0.1mpa
‑
0.3mpa,搅拌反应,持续通入环氧乙烷;
[0031]
(4)当环氧乙烷通入量为上述精制工艺制得的月桂醇精制品的9.0
‑
10.0摩尔当量时,停止加热,氮气置换、排空2
‑
5次优选3次,排除体系中的环氧乙烷;
[0032]
(5)重新通入氮气增压至0.1
‑
0.5mpa,优选0.20
‑
0.25mpa,继续保温130
‑
180℃,优选140
‑
155℃反应2
‑
10h,优选6h;
[0033]
(6)反应完毕,(例如用循环水)降温至70
‑
80℃,出料,得到聚桂醇粗品;
[0034]
(7)将所述聚桂醇粗品加入至1
‑
3倍体积优选2倍体积的乙腈中,搅拌溶解,加入0.2~1wt%,优选0.5wt%当量的活性炭,加热回流10
‑
50min,优选30min,趁热过滤,滤液减压浓缩至干,除去体系中的乙腈,得到剩余物;
[0035]
(8)将所述剩余物加入至离心桶中,置于离心机中在5000
‑
10000转/分钟的转速下离心分离,离心温度保持20
‑
30℃,离心分离10
‑
30min,优选20min,倒出上层清液,即为目标产品聚桂醇。
[0036]
优选地,上述聚桂醇的制备工艺中:
[0037]
步骤(1)中,上述精制工艺制得的月桂醇精制品:氢氧化钾二者摩尔比为1:(0.01~0.05),优选为1:0.02;和/或,
[0038]
步骤(1)中,所述氮气置换3次为:先通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压;和/或,
[0039]
步骤(4)中,所述环氧乙烷的通入量为,上述精制工艺制得的月桂醇精制品:环氧乙烷二者摩尔比为1:9.0~1:10.0;和/或,
[0040]
步骤(5)中,所述反应压力为0.2mpa,反应温度为140℃~150℃。
[0041]
第四方面,本发明提供上述聚桂醇的制备工艺所制备得到的聚桂醇。
[0042]
第五方面,本发明提供一种聚桂醇制剂,以上述聚桂醇的制备工艺所制备得到的聚桂醇为原料制备得到。
[0043]
聚桂醇可以加入本领域中的常规辅料,按照本领域中的常规工艺进行制备聚桂醇制剂,这是本领域技术人员所熟知的。
[0044]
本发明的有益效果:
[0045]
(1)本发明提供了一种尚没有文献记载的月桂醇的精制工艺,采用有机溶剂对月桂醇原料进行重结晶,最终制备得到了符合原料药要求的、单杂小于0.1%的月桂醇精制品,有效去除了月桂醇中可能引入的杂质,降低了月桂醇中的水分,而且该工艺路线简单,无需特殊设备,溶剂可回收利用,降低了工业生产成本;
[0046]
(2)本发明利用月桂醇精制工艺制备的月桂醇精制品作为原料制备聚桂醇,有效减少了聚桂醇中的杂质,减少了聚桂醇合成工艺中的脱水步骤,降低了能耗,因此适于工业化生产;所制备的聚桂醇不仅各项指标符合ep和中国药典要求,而且具备良好的耐氧化、耐高温能力,可在室温和空气存在条件下稳定保存,解决了现有聚桂醇产品需要低温(10℃以
下)、惰性气体(氮气、氩气等)保护下存放的问题,提高了聚桂醇的稳定性,不需要特殊包装进行存放,极大地降低了生产、储藏和运输成本,为聚桂醇制剂的生产提供了有效支持,必将大大降低患者的用药支出,造福于广大患者。通过本发明制备得到的聚桂醇制剂更容易符合制剂产品的要求。
具体实施方式
[0047]
下面结合具体实施例对本发明做进一步详细描述。
[0048]
本发明实施例中,月桂醇原料购自上海嘉辰化工有限公司,含量为99%;商品名为十二醇;
[0049]
四氢呋喃购自北京化学试剂公司,纯度为化学纯;
[0050]
乙腈购自北京化学试剂公司,纯度为化学纯。
[0051]
实施例1月桂醇精制品1的制备
[0052]
1)将1kg月桂醇原料、1kg四氢呋喃和1kg乙腈加入反应容器,升温至约28℃,搅拌至全溶,得到反应液。2)将反应液降温至0℃,搅拌析晶30分钟,抽滤,收集滤饼,得到中间体白色固体。3)将中间体白色固体放入反应容器中,缓慢升温至40℃,搅拌至融化,全融后减压蒸馏,蒸出体系中残留的有机溶剂,收集蒸馏产物,逐渐升温至90℃,控制真空度为
‑
0.1mpa,减压蒸馏4h,得到剩余产物,即为月桂醇精制品1。
[0053]
对实施例1制备的月桂醇精制品1按照进口药品注册标准聚桂醇注射液(jx20120149)中有关物质的要求进行检测,检测结果见表1。
[0054]
表1实施例1制备的月桂醇精制品1中有关物质的检测结果
[0055][0056]
实施例2月桂醇精制品2的制备
[0057]
1)将5kg月桂醇(原料)、5kg四氢呋喃和5kg乙腈加入反应容器,升温至约25℃,搅拌全溶,得到反应液。2)将反应液降温至
‑
2℃,搅拌析晶20分钟,抽滤,收集滤饼,得到中间体白色固体。3)将中间体白色固体放入至反应容器中,缓慢升温至20℃,搅拌至融化,全融后减压蒸馏,蒸出体系中残留的有机溶剂,收集蒸馏产物,逐渐升温至80℃,控制真空度≤
‑
0.1mpa,减压蒸馏5h,得到剩余产物,即为月桂醇精制品2。
[0058]
对实施例2制备的月桂醇精制品2按照进口药品注册标准聚桂醇注射液(jx20120149)中有关物质的要求进行检测,检测结果见表2。
[0059]
表2实施例2制备的月桂醇精制品2中有关物质的检测结果
[0060][0061]
实施例3月桂醇精制品3的制备
[0062]
1)将10kg月桂醇(原料)、10kg四氢呋喃和20kg甲苯加入反应容器,升温至30℃,搅拌全溶,得到反应液;2)将反应液降温至
‑
1℃,搅拌析晶30min,抽滤,收集滤饼,得到中间体白色固体。3)将中间体白色固体放入反应容器中,缓慢升温40℃,搅拌融化,全融后减压蒸馏,收集蒸馏产物,逐渐升温至70℃,控制真空度≤
‑
0.1mpa,减压蒸馏6h,得到剩余产物,即为月桂醇精制品3。对实施例3制备的月桂醇精制品3按照进口药品注册标准聚桂醇注射液(jx20120149)中有关物质的要求进行检测,检测结果见表3。
[0063]
表3实施例3制备的月桂醇精制品3中有关物质的检测结果
[0064][0065]
实施例4
[0066]
与实施例1同样地进行,只是重结晶溶剂为2kg四氢呋喃代替1kg四氢呋喃和1kg乙腈,产品检测结果如下:
[0067]
表4实施例4制备的月桂醇精制品4中有关物质的检测结果
[0068][0069][0070]
实施例5
[0071]
与实施例1同样地进行,只是重结晶溶剂为2kg甲苯代替1kg四氢呋喃和1kg乙腈,产品检测结果如下:
[0072]
表5实施例5制备的月桂醇精制品5中有关物质的检测结果
[0073][0074]
实施例6聚桂醇的制备
[0075]
以实施例1制备的月桂醇精制品1为原料制备聚桂醇,该制备方法包括以下步骤:
[0076]
(1)向高压反应釜中,加入实施例1制备的月桂醇精制品和0.005摩尔当量的氢氧化钾,锁紧釜盖,搅拌10min,转速为12rpm,通入氮气至0.2mpa,试漏,升温至90℃,氮气置换3次(即:先通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压),确保体系中空气置换充分。(2)通入氮气增压至0.15mpa,随后缓慢通入环氧乙烷增压至0.22mpa。(3)升温至145℃,压力保持在0.2mpa,搅拌反应,持续通入环氧乙烷。(4)当环氧乙烷通入量为月桂醇的9.0摩尔当量时,停止加热,氮气置换、排空三次,排除体系中的环氧乙烷。(5)重新通入氮气增压至0.2mpa,继续保温150℃反应6h。(6)反应完毕,用循环水降温至75℃,出料,得到聚桂醇粗品。(7)将得到的聚桂醇粗品加入2倍体积的乙腈中,搅拌溶解,加入0.3wt%的活性炭,加热回流30min,趁热过滤,滤液减压浓缩至干,除去体系中的乙腈,得到剩余物。(8)将剩余物加入至离心桶中,置于离心机中在8000转/分钟的转速下离心分离,温度保持25℃,离心分离20分钟,倒出上层清液,即为目标产品聚桂醇。
[0077]
实施例7聚桂醇的制备
[0078]
以实施例2制备的月桂醇精制品为原料制备聚桂醇,该制备方法包括以下步骤:
[0079]
(1)向高压反应釜中,加入实施例2制备的月桂醇精制品和0.03摩尔当量的氢氧化钾,锁紧釜盖,搅拌10min,转速为15rpm,通入氮气至0.5mpa,试漏,升温至80℃,氮气置换3次(即:先通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压),确保体系中空气置换充分。(2)通入氮气增压至0.15mpa,随后缓慢通入环氧乙烷增压至0.25mpa。(3)升温至140℃,压力保持在0.1mpa,搅拌反应,持续通入环氧乙烷。(4)当环氧乙烷通入量为月桂醇的9.5摩尔当量时,停止加热,氮气置换、排空三次,排除体系中的环氧乙烷。(5)重新通入氮气增压至0.25mpa,继续保温155℃反应4h。(6)反应完毕,用循环水降温至80℃,出料,得到聚桂醇粗品。(7)将得到的聚桂醇粗品加入2倍体积的乙腈中,搅拌溶解,加入0.5wt%当量的活性炭,加热回流30min,趁热过滤,滤液减压浓缩至干,除去体系中的乙腈。(8)将剩余物加入至离心桶中,置于离心机中在10000转/分钟的转速下离心分离,温度保持30℃,离心分离25分钟,倒出上层清液,即为目标产品聚桂醇。
[0080]
实施例8聚桂醇的制备
[0081]
以实施例3制备的月桂醇精制品为原料制备聚桂醇,该制备方法包括以下步骤:
[0082]
(1)向高压反应釜中,加入实施例3制备的月桂醇精制品和0.05摩尔当量的氢氧化
钾,锁紧釜盖,搅拌10min,转速为18rpm,通入氮气至0.5mpa,试漏,升温至90℃,氮气置换3次(即:先通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压,再通入氮气至正压,再抽真空至负压),确保体系中空气置换充分。(2)通入氮气增压至0.15mpa,随后缓慢通入环氧乙烷增压至0.20mpa。(3)升温至140℃,压力保持在0.1mpa,搅拌反应,持续通入环氧乙烷。(4)当环氧乙烷通入量为月桂醇的10.0摩尔当量时,停止加热,氮气置换、排空三次,排除体系中的环氧乙烷。(5)重新通入氮气增压至0.20mpa,继续保温150℃反应4h。(6)反应完毕,用循环水降温至80℃,出料,得到聚桂醇粗品。(7)将得到的聚桂醇粗品加入2倍体积的乙腈中,搅拌溶解,加入0.7wt%当量的活性炭,加热回流30min,趁热过滤,滤液减压浓缩至干,除去体系中的乙腈。(8)将剩余物加入至离心桶中,置于离心机中在8000转/分钟的转速下离心分离,温度保持30℃,离心分离25分钟,倒出上层清液,即为目标产品聚桂醇。
[0083]
对比例1对比样品1的制备
[0084]
参照公开号为cn1762948a的中国专利申请的实施例1制备聚桂醇。
[0085]
对比例2对比样品2的制备
[0086]
参照公开号为cn103922901a的中国专利申请的实施例1制备聚桂醇。
[0087]
对比例3对比样品3的制备
[0088]
参照公开号为cn104649863a的中国专利申请的制备例1和实施例1制备聚桂醇。
[0089]
实验例1样品质量对比
[0090]
分别对实施例6、实施例7、对比例1、对比例2和对比例3制备的聚桂醇按照注射液进口注册标准和ep标准中的要求进行检测,具体结果如表6所示。
[0091]
表6实施例6、实施例7、对比例1、对比例2和对比例3制备的聚桂醇的检测结果
[0092]
[0093][0094]
由表6可知,与对比例1
‑
3制备的聚桂醇相比,实施例6
‑
8制备的聚桂醇中游离月桂醇的含量更低、游离聚乙二醇的含量更低、有关物质杂质更低、含水量更少,杂质的降低,提高了产品的质量,更有利于聚桂醇作为注射液原料的使用,尽可能避免杂质导致的副作用的产生。
[0095]
实验例2稳定性试验
[0096]
以美国药典聚桂醇对照品(聚氧乙烯醚n=9,美国usp标准品)为对照品1,以中国食品药品检定研究院聚桂醇对照品为对照品2,对实施例6和实施例7制备的聚桂醇进行稳定性实验(包括60℃下加速试验前有关物质的检测、60℃下加速试验5天、10天、30天和60天有关物质的检测)。为了迅速了解样品的稳定性情况,对样品在60℃下进行加速试验,容器正常密封,无惰性气体保护。具体实验结果表7所示。
[0097]
表7实施例6
‑
7和对照品1
‑
2的稳定性试验结果
[0098][0099]
由表7可知,(1)实施例6
‑
7制备的聚桂醇和对照品1(usp对照品)均比对照品2(中检院对照品)更稳定,对照品2(中检院对照品)在60℃加速5天条件下迅速分解出大量未知杂质;
[0100]
(2)实施例6
‑
7制备的聚桂醇的稳定性明显优于对照品1(usp对照品)的稳定性,对照品1(usp对照品)在60℃加速10天时已产生了新的接近于1%的杂质,导致无法满足注射液对于原料药的要求;
[0101]
(3)实施例6
‑
7制备的聚桂醇在60℃加速60天下依然保持稳定,无明显新增杂质,确保了所制备的聚桂醇原料药可在室温下稳定存储,保证了未来用药的安全,降低了储存和运输成本。
[0102]
本发明的具体实施方式已得到详细的表述,应理解为这些实施例不仅用于例证的目的,不限于本发明的范围,同时本领域普通技术人员根据本发明所做的显而易见的改变和修饰也包含在本发明范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

