1.本发明属于有机化学和光电材料领域,具体涉及一类含苯乙烯基的菲并咪唑发光材料及其制备方法和应用。
背景技术:
2.以有机发光材料为蓝光/紫外led下转换材料可制备无机/有机杂化的白光led器件,这种器件与全无机led器件相比,由于其可以一定程度上减少对稀土金属材料的依赖而引起本领域技术人员的广泛关注。
3.有机蓝绿光材料是既发蓝光又可以发绿光的一类材料。将有机蓝绿光材料应用于有机/无机杂化白光led器件可以提高器件显色指数。菲并咪唑衍生物凭借着合成简单、成本低、收率高、量子产率高成为了使用频率非常高的发光材料,另外该类化合物可以用“一锅法”制备。通过苯乙烯基桥接菲并咪唑与其他发光基团(三苯胺、咔唑等),可以实现延长分子共轭从而构建蓝绿光材料,但是构建含苯乙烯基的菲并咪唑类发光材料,文献中通常采用碳碳偶联反应构建,反应过程中无法避免对贵重金属催化剂的使用,无法利用明显的特征官能团监测反应进程(journal of physical organic chemistry, 2017, 30 (12) e3695.)。因此,设计合成出结构新颖、合成过程避免贵重金属催化剂使用并易于监测反应进程、能应用于紫外led芯片激发的含苯乙烯基的菲并咪唑类发光材料极为重要。
技术实现要素:
4.本发明的目的之一是提供一类新型的含苯乙烯基的菲并咪唑类化合物,该类化合物可以作为有机发光材料应用于紫外led芯片激发构建有机/无机杂化的led发光器件。
5.本发明的目的之二在于提供一类合成苯乙烯基的菲并咪唑类化合物的制备方法。
6.本发明的目的之三在于提供上述化合物的应用。
7.一类含苯乙烯基的菲并咪唑类发光材料,其化学结构如下:
8.其中,r1基团为三苯胺或咔唑衍生物;r2基团选自氢、甲基、甲氧基、叔丁基、卤素、卤素取代的甲基中的一种。进一步的改进,所述三苯胺或咔唑衍生物为ⅱ所示结构中的任意一种:
一种上述含苯乙烯基的菲并咪唑类发光材料的制备方法,其特征在于,通过 wittig
‑
honor反应、还原反应、debus
‑
radziszewski“一锅法”制得含苯乙烯基的菲并咪唑类化 合物,此过程仅需通过三步法:合成路线如下:所述含苯乙烯基的菲并咪唑类发光材料的合成步骤如下:步骤一、将取代基(r1)甲醛(羰基类化合物):(4
‑
氰苄基)膦酸二乙酯:叔丁醇钾以1
‑
10.4: 1.2
‑
11.6:1.5
‑
10.4的摩尔比在溶剂中溶解进行反应,通过薄层层析的方法监测反应完成后通 过柱层析色谱对反应产物进行提纯,得到化合物1;步骤二、惰性气体氛围条件下,称取化合物1,置于三口烧瓶中,反应体系控制在
‑
77~
‑
22℃ 下加入二异丁基氢化铝溶液;化合物1与二异丁基氢化铝的摩尔比为1
‑
2:2.5
‑
7.5;通过薄层 层析监测反应完成,加入hcl溶液调节ph值,使ph值至1~5,随后采用乙酸乙酯萃取,合 并乙酸乙酯,干燥、旋蒸回收乙酸乙酯,得到化合物2;步骤三、称取化合物2、菲醌、苯胺或苯胺衍生物,醋酸铵和醋酸混合在三口烧瓶中形成反 应体系并采用惰性气体保护,然后加热升温到反应体系回流,通过薄层层析方法监
测反应终 点后停止加热,反应体系温度恢复至室温后加入去离子水,将溶液倒入分液漏斗并加入乙酸 乙酯萃取溶液,反复萃取水相直至水相澄清,合并乙酸乙酯萃取液后进行柱层析色谱分离得 到含苯乙烯基的菲并咪唑类发光材料;其中化合物2、菲醌、苯胺或苯胺衍生物和醋酸铵摩 尔比为1:1
‑
1.2:1:16。
9.进一步的改进:所述薄层层析的方法通过羰基和氰基在红外光谱中特征吸收峰的消失与再现,实现监测反应进程。
10.一种上述含苯乙烯基的菲并咪唑类发光材料的用途,所述含苯乙烯基的菲并咪唑类发光材料用于紫外光能激发的有机/无机杂化led器件中。
11.进一步的改进,所述紫外光的波长为300
‑
400 nm。
12.与现有技术相比,本发明具有如下优点和有益效果:本发明合成了一类含苯乙烯基的菲并咪唑类化合物,该类化合物的合成方法简单,通过三步反应即可得到目标产品,合成路径中涉及到了含甲酰基类化合物转化:第一步反应中原料甲酰基类化合物转化为含氰基的中间体;第二步反应中将含氰基类中间体转化为含甲酰基类中间体;第三步,含甲酰基类中间体转化为咪唑类基团,实现目标产品含苯乙烯基的菲并咪唑类化合物的转化。上述反应步骤中,有以下两个重要特征:1)可以利用甲酰基与氰基在红外光谱中的特征红外吸收,监测反应进程;2)反应过程中没有使用贵重金属催化剂。与现有技术相比,反应容易监测,未使用贵重金属催化的碳碳偶联反应,具有很好的现实意义。另外,本发明中提供的一类含苯乙烯基的菲并咪唑类化合物可以作为有机发光材料直接应用于紫外激发的有机/无机杂化的白光led器件。
附图说明
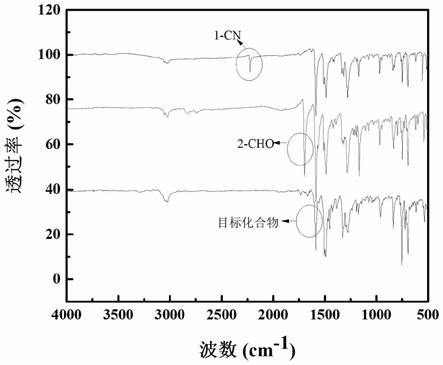
13.图1是本发明实施例一制备得到中间体与产品的红外光谱图。
14.图2是本发明实施例一中制备得到的e
‑1‑
(4
’‑
三苯胺基)
‑2‑
(4’,1
’’‑
苯基
‑4’’‑
苯基
‑
菲并咪唑)
‑
乙烯的化合物结构示意图、核磁氢谱(a)和核磁碳谱(b)图。
15.图3是本发明实施例一制备得到的e
‑1‑
(4
’‑
三苯胺基)
‑2‑
(4’,1
’’‑
苯基
‑4’’‑
苯基
‑
菲并咪唑)
‑
乙烯的质谱图。
16.图4是本发明实施例一基于e
‑1‑
(4
’‑
三苯胺基)
‑2‑
(4’,1
’’‑
苯基
‑4’’‑
苯基
‑
菲并咪唑)
‑
乙烯的制备得到有机/无机杂化led器件的光谱(a)和色坐标(b)图。
具体实施方式
17.为更好的理解本发明,现将具体实施方式列举如下,实施例可以对本发明做进一步的解释和说明,但本发明并不只局限于以下的实例。
18.(1)将取代基(r1)甲醛:(4
‑
氰苄基)膦酸二乙酯:叔丁醇钾比例为1
‑
4:1.2
‑
4.8:1.5
‑
3的原料加入到一定量的溶剂中,通过薄层层析的方法检测反应完成。通过柱层析色谱对上述粗产品进行提纯,得到液体或者固体产品,最大收率可达97%。
19.(2)惰性气体氛围条件下,称取(1)合成的淡黄色固体,置于三口烧瓶中,反应体系控制在
‑
77~
‑
22℃下加入物料比为1:1~1:12的1 mol/l~2.5 mol/l 浓度的二异丁基氢化铝(还原剂)甲苯溶液。通过薄层层析监测反应终点。加入约为10~30% hcl调节ph值,使ph值约为1~5。随后采用乙酸乙酯萃取,合并有机相,干燥溶剂后旋蒸回收溶剂,可得液体或者固体
(d, j = 16.3 hz, 1h)。
13
c nmr (101 mhz, cdcl3) δ 150.68, 147.60, 147.49, 138.91, 138.01, 137.55, 131.14, 130.23, 129.84, 129.54, 129.33, 129.28, 129.18, 129.13, 128.29, 128.25, 127.46, 127.30, 127.23, 126.28, 126.15, 126.07, 125.62, 124.87, 124.62, 124.13, 123.37, 123.17, 123.13, 123.06, 122.80, 120.87。红外光谱如图1(目标化合物)所示,其谱图中羰基的特征吸收峰消失,表明化合物2转化完成。生成的目标化合物的氢谱、碳谱如图2所示。其质谱如图3所示,表明结构正确。
25.本实施方式中原料4
‑
甲酰基三苯胺、叔丁醇钾、二异丁基氢化铝、苯胺和菲醌等为市售产品和利用公开方法制备产品均可,反应溶剂均为市售分析纯,所用的去离子水为实验室自制水。
26.本实施方式合成方法操作简单,原料价格低廉易得,后处理简单,反应路线中围绕特征官能团甲酰基、氰基的转变,容易通过红外检测。
27.本实施方式的合成路线如下所示:实施例2:e
‑1‑
(4
’‑
苯基
‑
咔唑基)
‑2‑
(4’,1
”‑
苯基
‑4”‑
苯基
‑
菲并咪唑)
‑
乙烯的制备(1)称取4
‑
(9h
‑
咔唑)苯甲醛1.0851g(4mmol),叔丁醇钾0.1683g(1.5mmol),二氯 甲烷40ml,(4
‑
氰苄基)膦酸二乙酯4ml(4.8mmol)于100ml三颈烧瓶中,采用薄层层 析跟踪反应到终点后加入去离子水,采用乙酸乙酯萃取3次,合并有机相后用无水硫酸镁干 燥24小时,过滤后采用柱层析分离,得到淡黄色固体0.8728g(2.3mmol),收率为58%。 外主要吸
收波数3000cm
‑1、2225cm
‑1、1450cm
‑1、1250cm
‑1、550cm
‑1。1h nmr(300mhz, cdcl3)δ8.15(d,j=7.3hz,2h),7.83
–
7.50(m,8h),7.43(d,j=5.3hz,4h),7.36
–
7.08(m, 4h)。
28.(2)称取(1)中淡黄色固体化合物0.8 g(2 mmol),甲苯30 ml于100 ml圆底烧瓶中,控制反应体系温度在
‑
77 ℃,氩气条件下,加入2.5 mol/l的二异丁基氢化铝3 ml,反应8小时,停止反应,用30%的盐酸调节ph至1,过滤,干燥后得到黄色固体0.6356 g,收率为80 %。红外主要吸收波数1700 cm
‑1、1450cm
‑1、1200 cm
‑1、750 cm
‑1。1h nmr (300 mhz, cdcl3) δ 10.00 (s, 1h), 8.15 (d, j = 7.7 hz, 2h), 7.89 (d, j = 8.1 hz, 2h), 7.72 (dd, j = 19.6, 8.2 hz, 4h), 7.59 (d, j = 8.3 hz, 2h), 7.53
ꢀ–ꢀ
7.12 (m, 8h)。
29.(3)氩气条件下,称取(2)中黄色固体0.3777 g(1 mmol)、9,10
‑
菲醌0.2124 g(1 mmol)、乙酸铵,量取7.5 ml的醋酸混合在25 ml的三口烧瓶中。随后采用氩气保护上述反应体系。回流反应72 h后,待体系温度恢复至室温后加入去离子。将溶液倒入分液漏斗并加入适量的乙酸乙酯萃取溶液,反复萃取水相直至水相澄清。合并有机相后通过旋蒸回收有机相,得到油状物。采用无水乙醇进行固化,得到黄色固体得到黄色粉末0.6 g,收率为82%。1h nmr (400 mhz, cdcl3) δ 8.93 (d, j = 7.6 hz, 1h), 8.81 (d, j = 8.4 hz, 1h), 8.75 (d, j = 8.3 hz, 1h), 8.18 (d, j = 7.7 hz, 2h), 7.84
ꢀ–ꢀ
7.41 (m, 19h), 7.37
ꢀ–ꢀ
7.13 (m, 7h)。红外主要吸收波数为3000 cm
‑1、1700 cm
‑1、1500 cm
‑1、1450 cm
‑1、750 cm
‑1。
30.实施例3:(1)称取4
‑
甲酰基三苯胺0.274g(1mmol),叔丁醇钾0.1683g(3mmol),二甲基亚砜 20ml,(4
‑
氰苄基)膦酸二乙酯3ml(3.6mmol)于50ml三颈烧瓶中,采用薄层层析跟踪 反应到终点后加入去离子水,采用乙酸乙酯萃取3次,合并有机相后用无水硫酸镁干燥24小 时,过滤后采用柱层析分离,得到0.31g黄色粉末,收率为90%。
31.(2)称取(1)中黄色固体产品0.3412 g(1 mmol),甲苯20 ml于50 ml圆底烧瓶中,控制反应体系温度在
‑
20 ℃,氩气条件下,加入2.5 mol/l的二异丁基氢化铝1 ml,反应12小时,停止反应,用10%的盐酸调节ph至1,过滤,干燥后得到黄色固体0.30 g(反应路线中化合物2),收率为80 %。
32.(3)氩气条件下,称取(2)中黄色固体产品0.37 g(1 mmol),4
‑
叔丁基苯胺0.1118 g(1 mmol),菲醌0.2082 g(1 mmol),醋酸铵1.322 g(16 mmol),醋酸60 ml于100 ml圆底烧瓶中,回流反应48 h后,加入去离子水,采用乙酸乙酯萃取3次,合并有机相后用无水硫酸镁干燥24小时,过滤后采用柱层析分离,得到0.3133 g,收率为80%。
33.实施例4:(1)称取4
‑
二对甲苯胺基苯甲醛1.8339g(6mmol),叔丁醇钾1.6950g(6mmol),二 甲基亚砜20ml,(4
‑
氰苄基)膦酸二乙酯6ml(6.6mmol)于50ml三颈烧瓶中,采用薄层 层析跟踪反应到终点后加入去离子水,采用乙酸乙酯萃取3次,合并有机相后用无水硫酸镁 干燥24小时,过滤后采用柱层析分离,得到1.05g黄色粉末,收率为86%。
34.(2)称取(1)中黄色固体产品0.8063 g(1 mmol),甲苯20 ml于50 ml圆底烧瓶中,氩气条件下,控制反应体系温度在
‑
30 ℃,加入2.5 mol/l的二异丁基氢化铝3 ml,反应12小时,停止反应,用10%的盐酸调节ph至2,过滤,干燥后得到黄色固体0.65 g(反应路线中化合物2),收率为80 %。
35.(3)氩气条件下,称取(2)中黄色固体产品0.4051 g(1 mmol),4
‑
氟苯胺0.1338 g(1.2 mmol),菲醌0.2082 g(1 mmol),醋酸铵1.3221 g(16 mmol),醋酸60 ml于100 ml圆底烧瓶中,回流反应48 h后,加入去离子水,采用乙酸乙酯萃取3次,合并有机相后用无水硫酸镁干燥24小时,过滤后采用柱层析分离,得到0.5123 g,收率为73%。
36.实施例5:(1)称取4
‑
(9h
‑4’4’‑
二叔丁基咔唑)苯甲醛3.5g(9mmol),叔丁醇钾1.1g(9mmol), 二甲基亚砜180ml,(4
‑
氰苄基)膦酸二乙酯9ml(9.6mmol)于500ml三颈烧瓶中,采用 薄层层析跟踪反应到终点后加入去离子水,采用乙酸乙酯萃取3次,合并有机相后用无水硫 酸镁干燥24小时,过滤后采用柱层析分离,得到3.2g淡黄色粉末,收率为73%。
37.(2)称取(1)中淡黄色固体产品0.96 g(2 mmol),甲苯20 ml于50 ml圆底烧瓶中,控制反应体系温度在
‑
50 ℃,氩气条件下,加入2.5 mol/l的二异丁基氢化铝2.4 ml,反应12小时,停止反应,用10%的盐酸调节ph至3,过滤,干燥后得到黄色固体0.69 g(反应路线中
nm)上,随后在干燥箱中以60 ℃烘1 h,130 ℃下固化2 h。固化完成后采用远方led光电测试系统测试器件性能,器件光效可达10 lm/w。
45.称取实施例3中的含苯乙烯基的菲并咪唑类产品0.15 g,将其置于总质量为1.5 g的硅胶封装胶水中(其中主胶a为0.3 g,固化剂b组分为1.2 g)。搅拌均匀后再超声30 min除去体系中的气泡。将含发光材料的硅胶胶水点胶于贴片led芯片(1 w,未封装半成品,395 nm)上,随后在干燥箱中以60 ℃烘1 h,130 ℃下固化2 h。固化完成后采用远方led光电测试系统测试器件性能,器件光效可达13 lm/w。
46.称取实施例4中的含苯乙烯基的菲并咪唑类产品0.1 g,将其置于总质量为1.5 g的硅胶封装胶水中(其中主胶a为0.3 g,固化剂b组分为1.2 g)。搅拌均匀后再超声30 min除去体系中的气泡。将含发光材料的硅胶胶水点胶于贴片led芯片(1 w,未封装半成品,300 nm)上,随后在干燥箱中以60 ℃烘1 h,130 ℃下固化2 h。固化完成后采用远方led光电测试系统测试器件性能,器件光效可达10 lm/w。
47.称取实施例5中的含苯乙烯基的菲并咪唑类产品0.25 g,将其置于总质量为1.5 g的硅胶封装胶水中(其中主胶a为0.3 g,固化剂b组分为1.2 g)。搅拌均匀后再超声30 min除去体系中的气泡。将含发光材料的硅胶胶水点胶于贴片led芯片(1 w,未封装半成品,380 nm)上,随后在干燥箱中以60 ℃烘1 h,130 ℃下固化2 h。固化完成后采用远方led光电测试系统测试器件性能,器件光效可达17 lm/w。
48.称取实施例6中的含苯乙烯基的菲并咪唑类产品0.27 g,将其置于总质量为1.5 g的硅胶封装胶水中(其中主胶a为0.3 g,固化剂b组分为1.2 g)。搅拌均匀后再超声30 min除去体系中的气泡。将含发光材料的硅胶胶水点胶于贴片led芯片(1 w,未封装半成品,365 nm)上,随后在干燥箱中以60 ℃烘1 h,130 ℃下固化2 h。固化完成后采用远方led光电测试系统测试器件性能,器件光效可达15 lm/w。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

