一种疫苗佐剂mpla的中间体、合成及应用
技术领域
1.本发明涉及一种疫苗佐剂mpla的中间体、合成及应用。
背景技术:
2.mpla是来自于革兰氏阴性细菌细胞壁中的内毒素(lps)的最内层脂质体(lipid a)部分,单磷酰脂质a(monophosphoryl lipid a,以下简称mpl)。lipid a是一个两亲结构,也是革兰氏阴性细菌具有毒性和免疫原性的关键结构,能引起机体的免疫反应。因此可将其作为佐剂添加在疫苗中以增加疫苗的免疫原性;一般来说,减毒活疫苗虽然免疫原性较强,但毒性也较大,可能会有致病风险;而无毒性的灭活疫苗,免疫原性较弱。佐剂的添加,可增强灭活疫苗的免疫原性,且不会对体质较弱者带来致病风险。同时减少抗原的使用量,增加疫苗供应量。mpl为铝盐之外的首个fda通过的人用新型免疫佐剂物质。mpl作用于toll-like受体4(tlr4),具有强免疫原性、机制明确、毒性小的优点。而现有的铝盐佐剂机制不明确,在注射部位发生红肿,疼痛的概率相比mpl大,且铝盐佐剂无法产生特异性细胞免疫。已有多个采用mpl佐剂系统的新疫苗品种在国内外获得上市,如乙肝疫苗fendrix,宫颈癌疫苗cerivix,带状疱疹疫苗shingrix以及疟疾疫苗mosquirix。并有二十余个处于临床阶段。
3.由于来源于不同细菌或同一细菌不同血清型的mpl其结构不同,基本差别在于脂肪链的数量/连接位置和脂肪链碳链长度,例如本专利合成的结构如下式所示;
[0004][0005]
n1和n3独立地为10、12或14,n2和n4独立地为8、10或12。
[0006]
现有mpl来源依赖生物发酵提取,成本昂贵,且提取过程中,脂多糖糖类异质性问题难以解决,极易引发纯度问题,带来使用安全隐患。而基于特殊理化性质及复杂结构,化学方法合成mpl极为困难,难以替代生物发酵。已有的mpl及其lipid a类似物的全合成报道也仅限于毫克级别,通常提供磷酸基团的配体一般为焦磷酸苄酯(di-o-benzyloxy(n,n-diisopropylamino)phosphine)或邻亚二甲苯基n,n-二乙基亚磷酰胺(n,n-diethyl-1,5-dihydro-3h-2,4,3-benzodioxaphosphepin-3-amine)作为磷酸基的来源,使用bn为永久性保护基。以上这些保护基最后都必须使用氢化条件才能脱除。例如现有文献制备类似的lipid a及其衍生物时,使用苄基,需要用pd/c在大气压或氢气加压条件下反应10-20 h,杂
质多,收率在45%-68%之间。且纯化方式复杂,有提到需要使用再生纤维素过滤并超声除去催化剂;再使用deae-纤维素离子交换树脂分离,需要使用多个不同组分的混合溶剂作为流动相冲出产物。再反复分液浓缩得到终产物。(见ref 1:alla zamyatina,harald sekljic,helmut brade.synthesis andpurity assessment of tetra-and pentaacyl lipid a of chlamydiacontaining(r)-3-hydroxyicosanoic acid. tetrahedron 60(2004)12113
–
12137。ref 2:kaustabh k.maiti,michael decastro,abu-baker m.abdel
-ꢀ
aal el-sayed.chemical synthesis and proinflammatory responses of monophosphoryl lipid a adjuvantcandidates.eur.j.org.chem.2010,80
–
91)。
[0007]
目前mpl类似物的全合成报道的合成路线均较长;除了使用的苄基保护基在后期脱保护时难以完全除去之外;为避免反应中产生副反应,通常需要一些临时性的保护基,导致涉及多个保护基,需要反复保护脱保护;并且在已报道的文献中四个脂肪链的链接连接次序各有不同,或在糖苷化后再连接四个脂肪链(0 4),或3 1的方法,脂肪链链接连接选择性不强,或位阻较大,糖苷化时选择性较低等问题。([1]j.am.chem.soc.2007,129,5200-5216;[2]wo 2013072768;[3]the chemical record,vol 6,333-343(2006))
[0008]
由此可见,在目前的文献报道中,氢化的结果并不理想,收率低,杂质多,反应时间长(一般大于20h),纯化条件繁琐复杂,难以规模化生产满足商业需求。
[0009]
因此,目前急需一种路线短,总收率较高的新的制备方法,从而能够大量合成mpla。
技术实现要素:
[0010]
本发明所要解决的技术问题是为了克服现有的疫苗佐剂mpla制备方法缺乏的缺陷,而提供了一种疫苗佐剂mpla的中间体、合成及应用。本发明合成的该中间体路线短,总收率明显增加。采用该mpla的关键中间体,在脱保护后即可得到mpla,为mpla的合成和放大提供了基础。
[0011]
本发明是通过下述技术方案来解决上述技术问题的。
[0012]
本发明提供了一种如式20所示的化合物的制备方法,其包括以下步骤:在有机溶剂和水的混合溶剂中,在碱存在下,将如式24所示的化合物进行如下所示的水解反应,得到所述的如式20所示的化合物即可;
[0013][0014]
所述的有机溶剂可为环醚类溶剂(例如四氢呋喃thf)。
[0015]
所述的有机溶剂与水的体积比可为1:1-1:2(例如1:1.2-1:1.5,又例如1:1.33)。
[0016]
所述的混合溶剂的用量可不做具体限定,以不影响反应即可;例如,所述的如式24所示的化合物与所述的混合溶剂的质量体积比可为10g/l-200g/l(例如50g/l-100g/l,又例如80g/l)。
[0017]
所述的碱可为碱金属氢氧化物(例如氢氧化锂、氢氧化钠和氢氧化钾中的一种或多种,又例如氢氧化锂)。所述的碱可为水溶液的形式。
[0018]
所述的碱与所述的如式24所示的化合物的摩尔比可为3:1-10:1(例如5:1-8:1)。
[0019]
所述的水解反应的温度可为50℃-100℃(例如所述的混合溶剂的共沸温度,又例如60℃-65℃)。
[0020]
所述的水解反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),一般以所述的如式24所示的化合物消失或含量不再减少时为反应终点;所述的水解反应的时间优选0.5
-ꢀ
12h(例如1h-5h)。
[0021]
所述的水解反应的后处理可为本领域常规的后处理,例如其包括以下步骤:所述的水解反应结束后,淬灭至ph值为7,有机溶剂稀释,洗涤,有机相干燥,过滤,浓缩,分离纯化,即可。所述的淬灭可采用饱和碳酸氢钠溶液。所述的淬灭至ph值为7可采用hcl水溶液(例如1.5m)。所述的有机溶剂稀释的有机溶剂可为卤代烷烃类溶剂(例如ch2cl2)。所述的洗涤可采用饱和碳酸氢钠溶液。所述的分离纯化优选柱层析分离,所述的柱层析分离的填料可为硅胶柱,所述的柱层析分离的洗脱剂可为石油醚与乙酸乙酯(例如石油醚:乙酸乙酯=5:1)。
[0022]
所述的如式20所示的化合物的制备方法,其还可包括如下步骤:在有机溶剂中,在硅试剂、还原剂和路易斯酸存在下,将如式23所示的化合物与2-萘甲醛进行如下所示的nap保护反应,得到所述的如式24所示的化合物即可;
[0023][0024]
所述的nap保护反应的操作和反应条件可为本领域该类nap保护反应中常规的操作和反应条件;本发明中优选如下:
[0025]
所述的nap保护反应中,所述的有机溶剂可为环醚类溶剂,所述的环醚类溶剂可为四氢呋喃 (thf)。
[0026]
所述的有机溶剂的用量可不做具体限定,以不影响反应即可;例如,所述的如式23所示的化合物与所述的有机溶剂的质量体积比可为5g/l-100g/l(例如10g/l-50g/l,又例如33g/l)。
[0027]
所述的硅试剂可为本领域该类反应中常规的硅试剂,其对化合物23的羟基进行硅烷基化保护;例如六甲基二硅氧烷(硅醚,(tms)2o)和/或三甲基氯硅烷,又例如六甲基二硅氧烷。
[0028]
所述的还原剂可为三乙基硅烷(et3sih)。
[0029]
所述的路易斯酸可为三氟甲磺酸酯,例如三甲基硅三氟甲磺酸酯(tmsotf)。
[0030]
所述的硅试剂与所述的如式23所示的化合物的摩尔比可为1:1-10:1(例如4:1-8:1,又例如6:1)。
[0031]
所述的还原剂与所述的路易斯酸的摩尔比可为3:1-10:1(例如4:1-6:1,又例如4.4:1)。
[0032]
所述的还原剂与与所述的如式23所示的化合物的摩尔比可为1:1-5:1(例如3:1-4:1,又例如3.5:1)。
[0033]
所述的2-萘甲醛与所述的如式23所示的化合物的摩尔比可为0.5:1-2:1(例如0.7:1-1:1,又例如 0.8:1)。
[0034]
所述的nap保护反应的温度可为-20℃至30℃(例如-10℃至10℃)。
[0035]
所述的nap保护反应中,可采用如下步骤:将所述的还原剂和路易斯酸加入到所述的如式23所示的化合物、2-萘甲醛与所述的有机溶剂的混合体系中,进行所述的nap保护反应。
[0036]
所述的nap保护反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),一般以所述的如式23所示的化合物消失或含量不再减少时为反应终点;所述的nap保护反应的时间优选0.5-12h(例如1h-5h)。
[0037]
所述的nap保护反应的后处理可为本领域常规的后处理,例如包括以下步骤:所述的nap保护反应结束后,有机溶剂稀释,洗涤,浓缩,分离纯化,即可。所述的有机溶剂稀释的有机溶剂可为卤代烷烃类溶剂(例如ch2cl2)。所述的洗涤可采用饱和碳酸氢钠溶液。所述的分离纯化优选柱层析分离或结晶,所述的结晶的溶剂可为石油醚与乙酸乙酯(例如石油醚:乙酸乙酯=5:1)。
[0038]
本发明提供了如下式20、式24所示的化合物,
[0039][0040]
本发明还提供了一种如上所示的如式24所示的化合物的制备方法,其包括以下步骤:在有机溶剂中,在硅试剂、还原剂和路易斯酸存在下,将如式23所示的化合物与2-萘甲醛进行如下所示的nap保护反应,得到所述的如式24所示的化合物即可;
[0041][0042]
所述的nap保护反应的操作和反应条件可为本领域该类nap保护反应中常规的操作和反应条件;本发明中优选如下:
[0043]
所述的nap保护反应中,所述的有机溶剂可为环醚类溶剂,所述的环醚类溶剂可为四氢呋喃thf。所述的有机溶剂的用量可不做具体限定,以不影响反应即可;例如,所述的如式23所示的化合物与所述的有机溶剂的质量体积比可为5g/l-100g/l(例如10g/l-50g/l,又例如33g/l)。
[0044]
所述的硅试剂可为本领域该类反应中常规的硅试剂,其对化合物23的羟基进行硅烷基化保护;例如六甲基二硅氧烷(硅醚,(tms)2o)和/或三甲基氯硅烷。
[0045]
所述的还原剂可为三乙基硅烷(et3sih)。
[0046]
所述的路易斯酸可为三氟甲磺酸酯(例如三甲基硅三氟甲磺酸酯tmsotf)。
[0047]
所述的硅试剂与所述的如式23所示的化合物的摩尔比可为1:1-10:1(例如4:1-8:1,又例如6:1)。
[0048]
所述的还原剂与所述的路易斯酸的摩尔比可为3:1-10:1(例如4:1-6:1,又例如
4.4:1)。
[0049]
所述的还原剂与与所述的如式23所示的化合物的摩尔比可为1:1-5:1(例如3:1-4:1,又例如3.5:1)。
[0050]
所述的2-萘甲醛与所述的如式23所示的化合物的摩尔比可为0.5:1-2:1(例如0.7:1-1:1,又例如 0.8:1)。
[0051]
所述的nap保护反应的温度可为-20℃至30℃(例如-10℃至10℃)。
[0052]
所述的nap保护反应中,可采用如下步骤:将所述的还原剂和路易斯酸加入到所述的如式23所示的化合物、2-萘甲醛与所述的有机溶剂的混合体系中,进行所述的nap保护反应。
[0053]
所述的nap保护反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),一般以所述的如式23所示的化合物消失或含量不再减少时为反应终点;所述的nap保护反应的时间优选0.5-12h(例如1h-5h)。
[0054]
所述的nap保护反应的后处理可为本领域常规的后处理,例如包括以下步骤:所述的nap保护反应结束后,有机溶剂稀释,洗涤,浓缩,分离纯化,即可。所述的有机溶剂稀释的有机溶剂可为卤代烷烃类溶剂(例如ch2cl2)。所述的洗涤可采用饱和碳酸氢钠溶液。所述的分离纯化优选柱层析分离或结晶,所述的结晶的溶剂可为石油醚与乙酸乙酯(例如石油醚:乙酸乙酯=5:1,体积比)。
[0055]
本发明还提供了一种所述的如式20所示的化合物在制备如式18所示的化合物中的应用;其可包括如下步骤:
[0056]
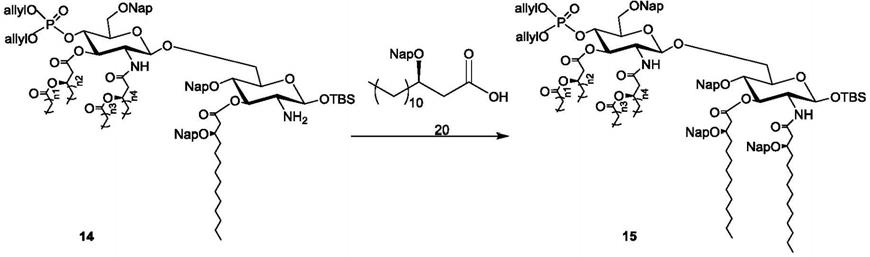
步骤(1)、在有机溶剂中,在缩合剂存在下,将如式14所示的化合物与如式20所示的化合物进行如下所示的酰胺化反应,得到所述的如式15所示的化合物即可;其中,n1和n3独立地为10、12 或14,n2和n4独立地为8、10或12;且当n2和n4为10时,n1和n3不同时为12;
[0057][0058]
步骤(2)、在有机溶剂中,在脱tbs保护剂存在下,将如式15所示的化合物进行如下所示的脱 tbs(tbdms,叔丁基二甲基)保护基反应,得到所述的如式16所示的化合物即可;
[0059]
[0060]
步骤(3)、在有机溶剂中,在碱、hcooh、pd催化剂及膦配体存在下,将如式16所示的化合物进行如下所示的脱allyl(烯丙基)保护基反应,得到所述的如式17所示的化合物即可;
[0061][0062]
步骤(4)、在有机溶剂中,将如式17所示的化合物与ddq(2,3-二氯-5,6-二氰对苯醌)进行如下所示的脱nap保护基反应,得到如式18所示的mpla化合物即可;
[0063][0064]
本发明中,通式中的为本领域常规的表示α构型、β构型,或α构型和β构型的混合物。
[0065]
在某一方案中,n1和n3独立地为10、12或14,n2和n4独立地为10;且n1和n3不同时为12。
[0066]
在某一方案中,n1和n3为10,n2和n4为10。
[0067]
在某一方案中,n1为10,n3为14,n2和n4为10。
[0068]
在某一方案中,n1为12,n3为10,n2和n4为10。
[0069]
步骤(1)中,所述的酰胺化反应的操作和反应条件可为本领域该类酰胺化反应中常规的操作和反应条件;本发明中优选如下:
[0070]
所述的酰胺化反应中,所述的有机溶剂可为卤代烷烃类溶剂,所述的卤代烷烃类溶剂可为二氯甲烷和/或三氯甲烷。所述的有机溶剂的用量可不做具体限定,以不影响反应即可;例如,所述的如式14 所示的化合物与所述的有机溶剂的质量体积比可为50g/l-200g/l(例如80g/l-100g/l)。
[0071]
所述的缩合剂可为1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc
·
hcl)、二环己基碳二亚胺(dcc)和n,n'-二异丙基碳二亚胺(dic)中的一种或多种,例如1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐。
[0072]
所述的缩合剂与所述的如式14所示的化合物的摩尔比可为1:1-10:1(例如4:1-8:1,又例如6:1)。
[0073]
所述的如式14所示的化合物与所述的如式20所示的化合物的摩尔比可为1:1-1:4(例如1:2-1:3)。
[0074]
所述的酰胺化反应的温度可为0℃至50℃(例如10℃-30℃)。
[0075]
所述的酰胺化反应可在惰性气体保护下进行,所述的惰性气体可为氮气或氩气。
[0076]
所述的酰胺化反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),一般以所述的如式14所示的化合物消失或含量不再减少时为反应终点;所述的酰胺化反应的时间优选 5-24h(例如10h-15h)。
[0077]
所述的酰胺化反应的后处理可为本领域常规的后处理,例如包括以下步骤:所述的酰胺化反应结束后,浓缩,分离纯化,即可。所述的分离纯化优选柱层析分离,所述的柱层析分离的洗脱剂可为甲苯与乙酸乙酯的混合溶液(例如甲苯/乙酸乙酯=10:1,体积比)。
[0078]
步骤(2)中,所述的脱tbs保护基反应的操作和反应条件可为本领域该类脱tbs保护基反应中常规的操作和反应条件;本发明中优选如下:
[0079]
所述的脱tbs保护基反应中,所述的有机溶剂可为环醚类溶剂,所述的环醚类溶剂可为四氢呋喃thf。所述的有机溶剂的用量可不做具体限定,以不影响反应即可;例如,所述的如式15所示的化合物与所述的有机溶剂的质量体积比可为5g/l-100g/l(例如10g/l-50g/l,又例如33g/l)。
[0080]
所述的脱tbs保护剂可为氢氟酸吡啶络合物(hf/pyridine,又称olah试剂,其中hf的质量百分浓度为65
–
70%);较佳地为氢氟酸吡啶络合物的吡啶溶液(例如3-6.5倍的吡啶稀释的氢氟酸吡啶络合物的溶液)。
[0081]
所述的脱tbs保护剂与所述的如式15所示的化合物的体积质量比可为1ml/g-20ml/g(例如3 ml/g-10ml/g)。
[0082]
所述的脱tbs保护基反应的温度可为-80℃至50℃(例如-40℃至30℃)。
[0083]
所述的脱tbs保护基反应中,可采用如下步骤:将所述的脱tbs保护剂加入到所述的如式15所示的化合物与所述的溶剂的混合体系中,进行如上述所述的脱tbs保护基反应。所述的加入温度可为-70℃至-30℃(例如-40℃)。加毕后,所述的脱tbs保护基反应的温度可为0℃-30℃。
[0084]
所述的脱tbs保护基反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),一般以所述的如式15所示的化合物消失或含量不再减少时为反应终点;所述的脱tbs保护基反应的时间优选1-24h(例如2h-12h)。
[0085]
所述的脱tbs保护基反应的后处理可为本领域常规的后处理,例如包括以下步骤:所述的脱tbs 保护基反应结束后,淬灭,有机溶剂萃取,干燥,过滤,浓缩,分离纯化,即可。所述的淬灭可采用饱和碳酸氢钠溶液。所述的萃取的有机溶剂可为卤代烷烃类溶剂(例如氯仿和/或二氯甲烷)。所述的分离纯化优选柱层析分离,所述的柱层析分离的填料可为c18填料,所述的柱层析分离的洗脱剂可依次为ch3cn,meoh/ea(例如meoh/ea=8:1,体积比)及meoh。
[0086]
步骤(3)中,所述的脱allyl保护基反应的操作和反应条件可为本领域该类脱allyl保护基反应中常规的操作和反应条件;本发明中优选如下:
[0087]
所述的脱allyl保护基反应中,所述的有机溶剂可为环醚类溶剂,所述的环醚类溶剂可为四氢呋喃(thf)。所述的有机溶剂的用量可不做具体限定,以不影响反应即可;例如,
所述的如式16所示的化合物与所述的有机溶剂的质量体积比可为1g/l-50g/l(例如10g/l-25g/l)。
[0088]
所述的脱allyl保护基反应中,所述的碱可为有机碱,所述的有机碱可为三乙胺(tea)和/或正丁基胺(n-bunh2)。
[0089]
所述的pd催化剂可为pd(pph3)4。
[0090]
所述的膦配体可为pph3。
[0091]
所述的pd催化剂的用量可为常规的催化剂量,例如所述的pd催化剂与所述的如式16所示的化合物的摩尔比可为0.1:1-0.5:1(例如0.2:1-0.4:1)。
[0092]
所述的膦配体与所述的pd催化剂的摩尔比可为2:1-5:1(例如4.4:1-4.75:1)。
[0093]
所述的碱与所述的pd催化剂的摩尔比可为20:1-50:1(例如42:1-45:1)。
[0094]
所述的hcooh与所述的pd催化剂的摩尔比可为20:1-50:1(例如42:1-45:1)。
[0095]
所述的脱allyl保护基反应的温度可为0℃至50℃(例如10℃-30℃)。
[0096]
所述的脱allyl保护基反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),一般以所述的如式16所示的化合物消失或含量不再减少时为反应终点;所述的脱allyl保护基反应的时间优选0.5-24h(例如1.5h-12h)。
[0097]
所述的脱allyl保护基反应的后处理可为本领域常规的后处理,例如包括以下步骤:所述的脱allyl 保护基反应结束后,浓缩,分离纯化,即可。所述的分离纯化优选柱层析分离,所述的柱层析的填料可为c18填料;所述的柱层析分离可包括如下步骤:使用含0.1%tea的ch3cn,含0.1%tea的 meoh,含0.1%tea的ch2cl2/meoh作为洗脱液依次进行洗脱。
[0098]
步骤(4)中,所述的脱nap保护基反应的操作和反应条件可为本领域该类脱nap保护基反应中常规的操作和反应条件;本发明中优选如下:
[0099]
所述的有机溶剂可为卤代烷烃类溶剂,所述的卤代烷烃类溶剂可为二氯甲烷和/或三氯甲烷。所述的有机溶剂的用量可不做具体限定,以不影响反应即可;例如,所述的如式17所示的化合物与所述的有机溶剂的质量体积比可为1g/l-50g/l(例如10g/l-20g/l)。
[0100]
所述的如式17所示的化合物与ddq的摩尔比可为1:2-1:220(例如1:4-1:20,又例如1:6-1:8)。由于每分子含有4当量nap,当ddq达到常规的用量(例如ddq与nap的摩尔比为1:1-2.25:1左右)时,即可高收率的得到化合物17。
[0101]
所述的脱nap保护基反应的温度可为10℃-50℃(例如20℃-30℃)。
[0102]
所述的脱nap保护基反应可在惰性气体保护下进行,所述的惰性气体可为氮气或氩气。
[0103]
所述的脱nap保护基反应较佳地在超声条件下进行,以加快反应进程。
[0104]
所述的脱nap保护基反应的进程可通过本领域常规的手段进行监控(例如tlc、hplc或lcms),一般以所述的如式17所示的化合物消失或含量不再减少时为反应终点;所述的脱nap保护基反应的时间优选0.5-6h(例如1h-1.5h)。
[0105]
所述的脱nap保护基反应的后处理可为本领域常规的后处理,例如包括以下步骤:所述的脱nap 保护基反应结束后,淬灭,浓缩,分离纯化,即可。所述的淬灭的溶剂可为胺类有机碱(例如三乙胺)。所述的分离纯化优选柱层析分离,所述的柱层析的填料可为c18填料;所述的柱层析分离可包括如下步骤:使用腈类溶剂(例如乙腈)除去过量ddq,再使用卤代烷烃类溶剂(例如二氯甲烷)和醇类溶剂(例如甲醇)的混合溶剂作为洗脱液进行洗脱;所
述的卤代烷烃类溶剂和醇类溶剂的体积比可为2:1
-ꢀ
4:1(例如3:1)。
[0106]
本发明中,如无特殊说明,室温指10-30℃;“h”是指小时;“过夜反应”是指反应8-16小时。
[0107]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0108]
本发明所用试剂和原料均市售可得。
[0109]
本发明的积极进步效果在于:本发明以nap为保护基,在后续操作中均可以方便的除去;合成的路线短,总收率明显增加。为mpla的合成和放大提供了基础。
具体实施方式
[0110]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0111]
化合物18-a系列实施例
[0112]
原料化合物1的制备
[0113][0114]
向加入2-脱氧-1-氧-(1,1-二甲基乙基)二甲基硅烷基-2-[(2,2,2-三氯乙氧基)羰基]氨基-3,4,6-三乙酰基-β-d-葡萄糖(10g,16.8mmol)的反应瓶中缓慢加入胍啶盐酸盐缓冲溶液(100ml,ph=8),室温搅拌 3.5h,tlc检测原料消耗完后,反应液用阳离子树脂中和,过滤浓缩,用二氯甲烷和饱和碳酸氢钠溶液萃取产物,收集有机层浓缩得到2-脱氧-1-氧-(1,1-二甲基乙基)二甲基硅烷基-2-[(2,2,2-三氯乙氧基) 羰基]氨基-β-d-葡萄糖(1-1,8.23g)。
[0115]
在反应瓶中将1-1与2-(二甲氧基甲基)-萘(5.1g,25mmol,1.5eq)溶解在50ml乙腈中,加入樟脑磺酸(0.39g,1.69mmol,0.1eq),室温下搅拌4h反应,加入三乙胺至中性,反应液用二氯甲烷与饱和碳酸氢钠溶液萃取,分液。有机相干燥旋干后得到黄色固体物。粗品过硅胶砂芯漏斗(pe:ea=5:1) 得到产物1(浅黄色固体,6.97g),两步收率为68.3%。
[0116]
化合物1:1h nmr(400mhz,cdcl3)δ7.96
–
7.50(m,7h),5.72(s,1h),5.17(d,j=6.3hz,1h), 4.88(d,j=7.7hz,1h),4.73(q,j=12.0hz,2h),4.36(dd,j=10.5,5.0hz,1h),4.13
–
4.01(m,1h),3.86 (t,j=10.3hz,1h),3.70
–
3.58(m,1h),3.52(td,j=9.7,5.0hz,1h),3.47
–
3.35(m,1h),2.96(s,1h),0.94 (d,j=8.2hz,9h),0.20
–
0.08(m,6h).
[0117]
13
c nmr(101mhz,cdcl3)δ154.54,134.52,133.78,132.87,128.41,128.29,127.75,126.69,126.41, 126.07,123.93,101.97,96.33,95.30,81.52,74.85,70.71,68.68,66.20,60.73,26.94,25.59,17.90,-4.14,
-ꢀ
5.26.
[0118]
原料化合物23的制备
[0119][0120]
参考如下参考文献中方法进行制备化合物23,并使用相同方法测定ee值:belma hasdemir,h
ü
lya onar,asymmetric synthesis of long chainβ-hydroxy fatty acid methyl esters as new elastase inhibitors.tetrahedron:asymmetry.(23)2012,1100-1105。文献标题不规范,已修改
[0121]
步骤(1)化合物22的制备
[0122]
将米氏酸(64.8g,0.45mol)和吡啶(48ml)溶于ch2cl2(100ml)中,在0℃下加入月桂酰氯(21,65.6g,0.3mol)。室温搅拌2.5小时。原料完全消耗后,用1m hcl(100ml)和水(100ml) 洗涤。有机层干燥过滤并浓缩。将其溶于甲醇(250ml)回流过夜。中间体消耗完后浓缩并通过al2o3柱色谱纯化(甲苯:乙酸乙酯=2∶1),得到淡黄色固体22(59.3g,77%)。
[0123]1h nmr(400mhz,cdcl3)δ3.70(3h,d,j=1.5),3.42(2h,s),2.50(2h,t,j=7.4),1.28(18h,s), 0.90(3h,t,j=6.6).
[0124]
13
c nmr(101mhz,cdcl3)δ202.51,167.58,137.67,128.96,128.15,125.25,52.02,48.84,42.90, 31.92,29.62,29.47,29.38,29.35,29.01,23.43,22.68,21.31,14.04.
[0125]
步骤(2)化合物23的制备
[0126][0127]
钌催化剂的制备:将180mg(r)-ru(oac)2(binap)(二乙酸根[(r)-( )-2,2'-二(二苯基膦基)-1,1'-联萘基]钌(ii),cas号:325146-81-4)溶于ch2cl2(5ml),加入1.42n hcl(0.35ml),室温下搅拌1 小时后旋干备用。
[0128]
将化合物22(15g)与制备好的钌催化剂溶于甲醇(50ml)。h2(1.5mpa)条件下65℃搅拌6 h。原料完全完后,将反应液浓缩并通过硅胶柱色谱法纯化(石油醚/乙酸乙酯=5∶1),得到化合物 23(白色固体,15g,98%,ee值=98.7%))。
[0129]1h nmr(400mhz,cdcl3)δ4.01(1h,dq,j=11.8,4.0),3.71(3h,s),2.90(1h,d,j=4.0),2.46(2 h,ddd,j=25.4,16.4,6.1),1.26(18h,s),0.88(3h,t,j=6.8).
[0130]
13
c nmr(101mhz,cdcl3)δ173.41,67.99,51.63,41.18,36.57,31.89,29.60,29.56,29.51,29.32, 25.47,22.65,14.05.
[0131]
对化合物23的制备进行反应条件筛选:
[0132]
底物与催化剂比例(s/c)与ee值的关系:以s/c=300催化剂量50℃大约4-6h反应完全,收率达到95%以上。为减少催化剂的使用,尝试了每20g底物,180mg催化剂的反应(s/c>300),收率仅为71%。
[0133]
将化合物22(20g)与制备好的180mg钌催化剂溶于甲醇(50ml)。h2(1.5mpa)条件下65℃搅拌6h。原料完全完后,将反应液浓缩并通过硅胶柱色谱法纯化(石油醚/乙酸乙酯=5∶1),得到化合物3(白色固体,10.7g,71%,ee值=92.3%)。
[0134]
对以上不同条件下的产物ee值进行分析,催化剂较少,收率下降的同时ee值也仅为92%。而 s/c=300的几批产物,ee值均在98%以上。
[0135]
实施例1化合物24的制备
[0136][0137]
将化合物23(10g,39mmol,1eq)与2-萘甲醛(18.14g,116mmol,3eq)溶于thf(100ml),冰浴下加入tmsotf(6.88g,31mmol,0.8eq),(tms)2o(37.68g,232mmol,6eq)和et3sih(15.7 g,135mmol,3.5eq)。0℃下反应1.5小时,将反应液用ch2cl2(150ml)稀释,并用饱和nahco3洗涤。将有机层旋干并重结晶(得到甲基萘,室温下是白色固体)(石油醚:乙酸乙酯=5:1),过滤以除去杂质(甲基萘)。收集滤液(含化合物24)并旋干,得到淡黄色油状液体的粗产物24,无需纯化直接用于下一步。1h nmr(400mhz,cdcl3)0.89(3h,t,j6.7),1.27
–
1.64(18h,m),2.35(2h,dd,j 8.4)2.49 (1h,dd,j15.0,5.3),2.62(1h,dd,j15.0,7.3),3.68(3h,s),3.88(1h,m),4.54(2h,m),7.25
–
7.35(7h,m).
[0138]
实施例2化合物20的制备
[0139][0140]
将实施例1中制备得到的化合物24溶于thf-h2o溶液(5:1,100ml),加入氢氧化锂水溶液 (9.41g,224mmol,94ml),回流12h。原料消失后,冷却至室温,加入1.5m hcl水溶液淬灭至 ph值为7。将混合物用ch2cl2(150ml)稀释,用饱和nahco3洗涤。有机相干燥并旋干。硅胶柱色谱法纯化(石油醚/乙酸乙酯=5∶1)得到化合物20(11.6g,两步收率77.9%,无色糖浆)。
[0141]1h nmr(400mhz,cdcl3)δ10.00(1h,s,oh),7.85(4h,dd,j=14.5,10.8),7.53(3h,d,j=4.2), 4.85
–
4.71(2h,m,napch2o),4.08
–
3.94(1h,m,h-3),2.76(1h,dd,j=15.2,6.9,h-2),2.64(1h,dd,j =15.2,4.3,h-2),1.84
–
1.58(2h,m,h-4),1.57
–
1.31(18h,m),1.00(3h,t,j=6.0).
[0142]
13
c nmr(101mhz,cdcl3)δ177.86,135.86,133.38,133.10,128.20,128.00,127.76,126.57,126.10, 125.99,125.89,75.94,71.71,39.81,34.37,32.05,29.77,29.71,29.48,25.27,22.82,14.26.
[0143]
实施例3化合物2的制备
[0144][0145]
将化合物19-a(5g,11.7mmol)与edc
·
hcl(2.25g,11.7mmol,1.2eq)溶于ch2cl2(50ml),室温搅拌15min后加入化合物1(5.93g,9.8mmol)和dmap(0.06g,0.5mmol,0.05eq),
室温下搅拌10 h反应完,反应液依次用ch2cl2(100ml)和饱和nahco3(60ml)洗涤。分液干燥浓缩后经硅胶柱色谱纯化(石油醚/乙酸乙酯=10:1)得到化合物2-a(8.6g,86.7%,无色糖浆)。tof
-ꢀ
ms:m/z:1036.46[m na]
。
[0146]
δ
h
(400mhz,cdcl3)7.91(1h,s),7.82(3h,dt,j 9.3,4.9),7.54(1h,d,j 8.5),7.50
–
7.44(2h,m), 5.65(1h,s,napch),5.40
–
5.29(2h,m,h-3,nh),5.17(1h,dd,j 12.2,6.2,lipid-h-3),4.88(1h,d,j 7.8, h-1),4.69(2h,dd,j 48.4,12.0,troc),4.34(1h,dd,j 10.5,4.9,h-6),3.84(1h,t,j 10.3,h-6),3.75(1h,t, j 9.4,h-4),3.62(1h,dd,j 16.8,7.7,h-2),3.54(1h,td,j 9.7,5.0,h-5),2.57(2h,ddd,j 20.5,15.2,6.3), 2.12(2h,t,j 7.4),1.56
–
1.41(4h,m),1.32
–
1.13(34h,m),0.92
–
0.85(15h,m),0.11(6h,d,j 8.6).
[0147]
δ
c
(101mhz,cdcl3)173.41,170.19,154.12,134.29,133.71,132.85,128.38,128.11,127.67,126.47, 126.16,125.81,123.73,101.76,96.82(c-1),95.31,78.95(c-4),74.66,71.14(c-3),69.98,68.72(c-6), 66.47(c-5),59.11(c-2),39.26,34.31,33.83,31.93,29.63,29.50,29.35,29.29,29.06,25.53,25.07,24.93, 22.69,17.88,14.13,-4.21,-5.29.
[0148]
实施例4化合物3-a的制备
[0149][0150]
将化合物2-a(2g,1.97mmol)溶于thf(20ml),依次在冰浴条件下加入bh3·
me3n(0.57g,7.69 mmol,3.9eq),alcl3(1.52g,11.4mmol),h2o(69mg,3.83mmol,1.9eq)反应液在室温下搅拌1.5h。加水(20ml)、1m hcl溶液(20ml)淬灭,反应液用ch2cl2(30ml)分液,干燥浓缩后经硅胶柱色谱纯化(石油醚/乙酸乙酯=10:1)得到化合物3-a(1.72g,86%,无色油状液体)。tof
-ꢀ
ms:m/z:1038.52[m na]
.
[0151]
δ
h
(400mhz,cdcl3)7.85
–
7.75(4h,m),7.46(3h,dd,j 7.7,4.0),5.37(1h,d,j 9.2,nh),5.15(1h, s,lipid-h-3),5.03(1h,t,j 9.7,h-3),4.81
–
4.71(4h,m,h-1,napch2,troc),4.63(1h,d,j 11.9,troc), 3.81(2h,qd,j 10.5,3.8,h-6),3.69(1h,t,j 9.8,h-4),3.65
–
3.54(2h,m,h-2,h-5),3.47(1h,s,oh), 2.62
–
2.48(2h,m,lipid-h-2),2.28(2h,t,j 7.3,lipid
’-
h-2),1.57(4h,s,lipid
’-
h-3,lipid-h-4),1.25(34h, s),0.88(15h,s),0.12(6h,d,j 17.5).
[0152]
δ
c
(101mhz,cdcl3)174.35,171.77,154.27,135.54,133.29,133.02,128.20,127.89,127.71,126.29, 126.11,125.88,125.56,96.50(c-1),75.91(c-3),74.72(c-5),74.61,73.73,70.96,70.19(c-4),70.01(c-6), 60.43,57.85(c-2),40.10,34.58,34.50,31.94,29.65,29.56,29.52,29.37,29.30,29.14,25.61,25.16,24.99, 22.71,21.06,
17.93,14.21,14.14,-4.06,-5.28.
[0153]
实施例5化合物4的制备
[0154][0155]
(1)
[0156]
将化合物3-a(0.5g,0.489mmol)与四氮唑(102mg,1.45mmol,3eq)溶于超干乙腈(10ml),加入烯丙基配体(己二烯-n,n-二异丙基亚磷酰胺,0.24g,0.978mmol,2eq),室温下反应40min后原料消耗完。
[0157]-40℃下将mcpba(211mg,1.22mmol,2.5eq)溶于超干二氯甲烷(15ml),加入上述反应体系,缓慢升至-10℃,40min反应完加饱和硫代硫酸钠溶液(20ml)淬灭,饱和碳酸氢钠洗涤(40ml),干燥旋干硅胶柱色谱纯化(石油醚:乙酸乙酯=8:1)得化合物4-a(0.53g,92%)。tof
-ꢀ
ms:m/z:1198.67[m na]
.
[0158]
δ
h
(400mhz,cdcl3)7.85
–
7.77(4h,m),7.50
–
7.44(3h,m),5.90
–
5.73(2h,m,ch2=ch-ch2o-), 5.41
–
5.16(6h,m,nh,lipid-h-3,ch2=ch-ch2o-),4.97(1h,d,j 7.9,h-1),4.79-4.63(4h,td,j 12.3,12.4,12.0,troc,nap-ch2),4.49
–
4.38(5h,m,h-4,ch2=ch-ch2o-),3.85(1h,d,j 9.6,h-6),3.75(1 h,d,j 5.4,h-6),3.72
–
3.65(1h,m,h-5),3.50
–
3.40(1h,m,h-2),2.64
–
2.60(2h,m),2.28(2h,t,j 7.4),1.58(4h,br,j 7.3),1.25(34h,s),0.91
–
0.86(15h,m),0.13(6h,d,j 19.2).
[0159]
δ
c
(101mhz,cdcl3)173.57,170.37,153.95,135.65,133.27,132.98,132.27,132.21,132.14,128.09, 127.87,127.68,126.23,126.05,125.81,125.65,118.56,118.40,95.63(c-1),95.32,74.59,74.12(c-5), 74.06(c-4),73.61,72.42(c-3),70.06,68.70(c-6),68.62,68.56,68.46,68.41,58.67(c-2),39.59,34.51,34.24, 31.92,29.66,29.64,29.57,29.52,29.35,29.20,25.61,25.17,25.07,22.69,22.57,17.93,14.12,-4.15,-5.25.
[0160]
(2)
[0161]
将化合物3-a(0.5g,0.489mmol)与四氮唑(102mg,1.45mmol,3eq)溶于超干乙腈(10ml),加入烯丙基配体(0.18g,0.73mmol,1.5eq),室温下反应2h后原料未消耗完,补加0.5eq烯丙基配体, 30min后原料消耗完。-40℃下将mcpba(211mg,1.22mmol,2.5eq)溶于超干二氯甲烷(15ml),加入反应体系,缓慢升至-10℃,30min反应完加饱和硫代硫酸钠溶液(20ml)淬灭,饱和碳酸氢钠洗涤(40ml),干燥旋干硅胶柱色谱纯化(石油醚:乙酸乙酯=8:1)得化合物4-a(0.49g,84.7%)。
[0162]
(3)
[0163]
将化合物3-a(0.5g,0.489mmol)与四氮唑(171mg,2.45mmol,5eq)溶于超干乙腈
(10ml),加入烯丙基配体(0.24g,0.978mmol,2eq),室温下反应40min后原料消耗完,-40℃下将mcpba(211 mg,1.22mmol,2.5eq)溶于超干二氯甲烷(15ml),加入反应体系,缓慢升至-10℃,40min反应完加饱和硫代硫酸钠溶液(20ml)淬灭,饱和碳酸氢钠洗涤(40ml),干燥旋干硅胶柱色谱纯化(石油醚:乙酸乙酯=8:1)得化合物4-a(0.474g,82.3%)。
[0164]
实施例6化合物5-a的制备
[0165][0166]
将化合物4-a(0.8g,0.68mmol)溶于thf(24ml),
–
40℃加入稀释于15ml吡啶的hf/pyridine (2.4ml,65
–
70%)。缓慢升至室温反应12h,反应液用nahco3(40ml)淬灭,加入ch2cl2(50ml)分液,干燥旋干硅胶柱色谱纯化(石油醚:乙酸乙酯=4:1)得化合物5-a(0.657g,91%,白色固体)。tof
-ꢀ
ms:m/z:1084.48[m na]
。
[0167]
δ
h
(400mhz,cdcl3)7.85
–
7.77(4h,m),7.51
–
7.44(3h,m),5.91
–
5.65(2h,m,ch2=ch-ch2o-), 5.61(1h,d,j 9.4,nh),5.42
–
5.26(3h,m,h-1,ch2=ch-ch2o-),5.23
–
5.09(4h,m,h-3,ch2=ch
-ꢀ
ch2o-,lipid-h-3),4.80-4.69(4h,m,troc,nap-ch2),4.45
–
4.40(3h,m,h-4,ch2=ch-ch2o-),4.36
–ꢀ
4.27(2h,m,ch2=ch-ch2o-),4.25
–
4.19(1h,m,h-5),4.01
–
3.94(1h,m,h-2),3.84
–
3.75(2h,m,h
-ꢀ
6),2.60(2h,ddd,j 21.4,16.2,6.3),2.24(2h,t,j 7.6),1.56(4h,d,j 7.0),1.25(34h,s),0.88(6h,t,j 6.8).
[0168]
δ
c
(101mhz,cdcl3)173.38,170.76,154.31,135.19,133.23,133.04,132.28,132.22,132.09,132.02, 128.23,127.92,127.70,126.67,126.15,125.95,125.81,118.66,118.38,95.37,91.50(c-1),74.65,73.63(c
-ꢀ
4),70.82(c-3),69.90,69.57,69.52(c-5),68.67,68.61,68.47,68.40,68.35(c-6),54.27(c-2),39.10,34.46, 34.15,31.92,29.64,29.57,29.53,29.36,29.32,29.18,25.18,25.01,22.69,14.12.
[0169]
实施例7化合物6-a的制备
[0170][0171]
将化合物5-a(1.3g,1.22mmol)溶于超干(dry)ch2cl2(20ml),n2保护下加入2,2,2-三氟-n-苯乙酰亚胺氯(1.52g,7.32mmol,6eq)与dbu(371mg,2.44mmol,2eq),室温反应30mins。将反应液浓缩柱色谱纯化(石油醚:乙酸乙酯=15:1with 0.1%et3n)得到化合物
6-a(1.11g,74%,无色糖浆)。 tof-ms:m/z:1255.48[m na]
。
[0172]
实施例8化合物7的制备
[0173][0174]
(1)
[0175]
将化合物1(3.39g,5.58mmol)与化合物20(4.29g,11.16mmol,2eq)加入反应瓶,加入40ml 二氯甲烷后,冰浴下加入edc
·
hcl(2.14g,11.16mmol,2eq)与dmap(135mg,1.1mmol,0.2eq),室温搅拌3h后化合物1消耗完。加入饱和碳酸氢钠溶液(25ml)洗涤分液,干燥过滤旋干后重结晶纯化(二氯甲烷:甲醇=1:12)得到化合物7(5.08g,93.5%)。tof-ms:m/z:994.41[m na]
。
[0176]
δ
h
(400mhz,cdcl3)7.81(1h,s),7.76
–
7.61(7h,m),7.48
–
7.35(5h,m),7.30(1h,d,j 8.4),5.50 (1h,s,napch),5.39(1h,t,j 9.9,h-3),5.24(1h,d,j 9.1,nh),4.84(1h,d,j 7.8,h-1),4.67
–
4.62(3h, m,napch
2,-och2ccl3),4.52(1h,d,j 11.8,-och2ccl3),4.31(1h,dd,j 10.4,4.7,h-6),3.85
–
3.81(1h, m,lipid-h-3),3.78(1h,d,j 10.3,h-6),3.71(1h,t,j 9.5,h-4),3.67
–
3.60(1h,m,h-2),3.53(1h,td,j 9.5,5.1,h-5),2.74(1h,dd,j 14.9,6.0,lipid-h-2),2.55(1h,dd,j 14.8,5.6,lipid-h-2),1.54(2h,m, lipid-h-4),1.27(18h,s),0.91(12h,s),0.13(6h,d,j 12.0).
[0177]
δ
c
(101mhz,cdcl3)171.82,154.36,136.11,134.41,133.85,133.41,133.13,132.99,128.53,128.28, 128.23,128.11,127.88,127.85,126.60,126.51,126.31,126.20,126.04,125.96,125.91,123.85,101.88,97.14 (c-1),79.18(c-4),75.73(c-6),74.88,71.39(c-3),71.28,68.91(c-6),66.76(c-5),60.64,59.29(c-2),39.77, 34.71,32.15,29.87,29.86,29.80,29.77,29.58,25.74,25.45,22.92,21.28,18.08,14.43,14.37,-3.98,-5.08.
[0178]
(2)
[0179]
将edc
·
hcl(1.087g,5.67mmol,1.2eq),化合物20(2.18g,5.67mmol,1.2eq)加入反应瓶,加入25ml二氯甲烷,室温搅拌15min后加入化合物1(2.87g,4.72mmol),dmap(29mg,0.237mmol, 0.05eq),反应12h化合物1未消耗完。加入饱和碳酸氢钠溶液(25ml)洗涤分液,干燥过滤旋干后柱层析纯化(石油醚:乙酸乙酯=7:1)得到化合物7(2.72g,59.23%)。
[0180]
(3)
[0181]
将edc
·
hcl(1.81g,9.44mmol,2eq),化合物20(2.54g,6.6mmol,1.4eq)加入反应瓶,加入 25ml二氯甲烷,室温搅拌15min后加入化合物1(2.87g,4.72mmol),dmap(29mg,0.237mmol, 0.05eq),反应12h化合物1未消耗完。加入饱和碳酸氢钠溶液(25ml)洗涤分液,干燥过滤旋干后柱层析纯化(石油醚:乙酸乙酯=7:1)得到化合物7(3.27g,71.2%)。
[0182]
(4)
[0183]
将edc
·
hcl(2.65g,13.8mmol,2eq),化合物20(5.33g,13.8mmol,2eq)加入反应瓶,加入50 ml二氯甲烷,室温搅拌15min后加入化合物1(4.12g,6.78mmol),dmap(170mg,1.39mmol,0.2 eq),反应12h化合物1消耗完。加入饱和碳酸氢钠溶液(50ml)洗涤分液,干燥过滤旋干后柱层析纯化(石油醚:乙酸乙酯=7:1)得到化合物7(5.22g,79.1%)。
[0184]
(5)
[0185]
将化合物1(3.39g,5.58mmol)与化合物20(4.29g,11.16mmol,2eq)加入反应瓶,加入40ml 二氯甲烷后,冰浴下加入edc
·
hcl(2.14g,11.16mmol,2eq)与dmap(135mg,1.1mmol,0.2eq),室温搅拌3h后化合物1消耗完。加入饱和碳酸氢钠溶液(25ml)洗涤分液,干燥过滤旋干后柱层析纯化(石油醚:乙酸乙酯=7:1)得到化合物7(4.71g,86.7%)。
[0186]
(6)
[0187]
将化合物1(3.39g,5.58mmol)与化合物20(4.29g,11.16mmol,2eq)加入反应瓶,加入40ml 二氯甲烷后,冰浴下加入edc
·
hcl(2.14g,11.16mmol,2eq)与dmap(135mg,1.1mmol,0.2eq),室温搅拌3h后化合物1消耗完。加入饱和碳酸氢钠溶液(25ml)洗涤分液,干燥过滤旋干后重结晶纯化(二氯甲烷:甲醇=1:8)得到化合物7(3.93g,72.4%)。
[0188]
实施例9化合物8的制备
[0189][0190]
化合物7(6g,6.16mmol)溶于二氯甲烷(60ml),加入醋酸(12ml,2v)和锌粉(6g,1v) (v是指质量当量),室温剧烈搅拌2h反应完,依次用饱和碳酸氢钠(60ml),饱和食盐水(60 ml)洗涤。分液干燥浓缩后得到化合物8(5.3g)。tof-ms:m/z:821.134[m na]
。直接用于下一步反应。
[0191]
实施例10化合物9的制备
[0192][0193]
(1)
[0194]
将实施例9中制备得到的化合物8溶于二氯甲烷(100ml),冰浴下加入fmoccl(3.2g,12.36 mmol,2eq),dipea(1.6g,12.36mmol,2eq),室温下搅拌2h反应完,加入饱和食盐水(100ml) 洗涤,分液干燥旋干后重结晶(二氯甲烷:甲醇=1:10)得到化合物9(5.6g,89.1%)。tof
-ꢀ
ms:m/z:1042.7[m na]
。
[0195]
δ
h
(400mhz,cdcl3)7.83(1h,s),7.78
–
7.65(7h,m),7.64
–
7.44(5h,m),7.40(6h,ddd,j 15.3, 9.7,5.3),7.32
–
7.24(3h,m),5.53(1h,s,napch),5.44(1h,t,j 9.4,h-3),
4.97(1h,d,j 8.7,nh),4.91 (1h,d,j 7.2,h-1),4.63(1h,d,j 11.8,fmoc-ch2),4.51(1h,d,j 11.8,,fmoc-ch2),4.32(1h,br,h-6), 4.28(2h,d,j 6.5,napch2),4.17(1h,d,j 6.4,,fmoc-ch),3.82(2h,s,h-6,lipid-h-3),3.78
–
3.71(1h, m,h-4),3.67(1h,d,j 8.9,h-2),3.58(1h,br,h-5),2.70(1h,dd,j 14.8,6.3),2.50(1h,dd,j 14.8,5.5), 1.55
–
1.37(2h,m),1.29
–
0.96(18h,m),0.91
–
0.81(12h,m),0.09(6h,d,j 13.5).
[0196]
δ
c
(101mhz,cdcl3)155.78,143.79,141.23,135.92,134.24,133.63,133.17,132.87,132.78,128.31, 128.05,127.92,127.86,127.63,127.04,126.34,126.26,126.06,125.93,125.81,125.69,125.15,123.64, 119.96,101.65,97.14(h-1),79.00(h-4),75.62(h-6),71.25(h-3),71.19,68.76(h-6),67.19,66.60(h-5), 58.98(h-2),47.04,39.74,34.53,31.92,29.61,29.53,29.34,25.49,25.16,22.69,17.86,14.14,-4.21,-5.36.
[0197]
(2)
[0198]
化合物7(6g,6.16mmol)溶于二氯甲烷(60ml),加入醋酸(12ml,2v)和锌粉(6g,1v),室温剧烈搅拌2h反应完,依次用饱和碳酸氢钠(60ml),饱和食盐水(60ml)。分液干燥浓缩后得到化合物8(5.3g)。将其溶于二氯甲烷(100ml),冰浴下加入fmoccl(3.2g,12.36mmol,2eq), dipea(1.6g,12.36mmol,2eq),室温下搅拌2h反应完,加入饱和食盐水(100ml)洗涤,分液干燥旋干后重结晶(甲醇,5v)得到化合物9(4.91g,78.1%)。
[0199]
实施例11化合物10的制备
[0200][0201]
将化合物9(1.5g,1.47mmol)与分子筛(1.5g)溶于超干ch2cl2(60ml),n2保护下
–
78℃加入三乙基硅烷(0.55ml,3.67mmol,2.5eq)与phbcl2(0.76ml,5.88mmol),反应液在
–
78℃搅拌1h。反应结束后加入甲醇(6ml)淬灭,三乙胺调ph~8。将反应液过滤后饱和nahco3(10ml)洗涤。干燥旋干硅胶柱色谱纯化(石油醚:乙酸乙酯=6:1)得到化合物10(1.2g,80.1%,无色油状液体)。tof
-ꢀ
ms:m/z:1044.45[m na]
。
[0202]
δ
h
(400mhz,cdcl3)7.75
–
7.73(1h,m),7.70(7h,dd,j 14.7,5.4),7.66(2h,d,j 5.3),7.62(2h,d, j 5.8),7.53(1h,d,j 3.9),7.44
–
7.38(6h,m),7.30
–
7.25(3h,m),5.32(1h,t,j 9.8,h-3),4.93(1h,d,j 9.3,nh),4.79(1h,d,j 6.6,h-1),4.73(2h,q,j 11.7,napch2),4.57(2h,dd,j 24.5,11.7,fmoc-ch2), 4.24(2h,d,j 7.1,lipid-napch2),4.17
–
4.10(1h,m,fmoc-ch),3.85-3.80(2h,s,h-6,lipid-h-3),3.74 (1h,d,j 3.9,h-4),3.73
–
3.69(1h,m,h-6),3.60(1h,dd,j 18.4,9.4,h-2),3.49(1h,d,j 8.3,h-5),2.55 (1h,dd,j 15.6,7.1),2.39(1h,dd,j 15.5,4.9),1.55
–
1.39(2h,m),1.22
–
1.06(18h,m),0.87
–
0.82(12 h,m),0.08(6h,d,j 13.4).
[0203]
δ
c
(101mhz,cdcl3)172.00,155.84,143.86,141.23,135.99,135.05,133.23,
133.15,132.98,132.92, 128.26,128.04,127.95,127.87,127.69,127.65,127.04,126.67,126.29,126.14,126.00,125.97,125.87, 125.79,125.74,125.22,119.96,96.66(h-1),75.72(h-4),75.28(h-5),74.64(h-3),71.47(fmoc-ch2),67.24 (lipid-napch2),62.00(h-6),58.48(h-2),53.46,47.06(fmoc-ch),39.74,34.19,31.96,29.67,29.65,29.62, 29.60,29.38,25.52,25.15,22.73,17.90,14.17,-4.07,-5.25.
[0204]
实施例12化合物11的制备
[0205][0206]
(1)
[0207]
在反应瓶中加入受体化合物10(606mg,0.593mmol),给体化合物6-a(1.1g,0.89mmol,1.5eq)分子筛(600mg),n2条件下加入超干ch2cl2(10ml),在-20℃下加入二氯甲烷稀释100倍的tfoh (10μl,0.12mmol,0.2eq),
–
20℃下搅拌40min;加入甲醇(5ml)淬灭,三乙胺调ph至8。反应液用 ch2cl2(35ml)和饱和nahco3(10ml)洗涤。有机相干燥浓缩,经柱层析纯化(pe:ea=10:1)得到糖苷化产物11-a(1.03g,84%)。tof-ms:m/z 2088.14[m na]
。
[0208]
δ
h
(400mhz,cdcl3)7.80
–
7.21(29h,m),5.89
–
5.68(2h,m,diallyl),5.48(1h,d,j 7.0,nh’),5.37 (1h,t,j 9.6,h-3’),5.32
–
5.10(6h,m,h-3,lipid-h-3,diallyl),4.87(1h,d,j 9.1,nh),4.80(1h,d,j 7.7, h-1’),4.74(1h,d,j 3.3,h-1),4.70(4h,d,j 12.8,troc,nap),4.64(2h,d,j 3.3,nap-ch2),4.53(2h,dd, j 29.1,11.9,fmoc),4.45(1h,s,h-4’),4.43
–
4.31(5h,m,h-4,diallyl),4.24(2h,d,j 6.8,acceptor-lipid
-ꢀ
nap),4.16
–
4.11(1h,m,fmoc-ch),4.07(1h,d,j 10.3,h-6),3.83(2h,d,j 10.2,h-6,acceptor-lipid-h
-ꢀ3’
),3.72(2h,dd,j 10.3,6.0,h-6’),3.65
–
3.54(3h,m,h-2,h-5,h-5’),3.41(1h,dd,j 17.7,8.4,h-2’), 2.61(2h,t,j 6.6),2.50(1h,dd,j 15.6,7.1),2.35(1h,dd,j 15.8,5.1),2.28(2h,t,j 7.4),1.59(6h,d,j 7.7),1.42(4h,ddd,j 14.5,10.8,6.1),1.32
–
1.21(52h,m),0.86(18h,dd,j 14.9,7.8),0.15
–
0.05(6h, m).
[0209]
δ
c
(101mhz,cdcl3)δ172.65,170.83,169.04,154.72,152.94,142.82,140.16,134.94,134.60,134.29, 132.19,132.15,132.08,131.91,131.84,131.82,131.29,131.22,131.13,131.06,129.85,127.80,127.12, 127.06,126.93,126.85,126.79,126.62,126.60,126.55,125.97,125.32,125.17,125.10,125.02,124.90, 124.85,124.78,124.70,124.67,124.61,124.18,118.86,117.44,117.27,99.00(c-1’),95.53(c-1),94.38, 75.08(c-5’),74.58(c-5)73.71(c-3),73.43(c-4’),73.31(c-4),73.11,73.05,72.96,72.91,72.60,71.37(c-3’), 70.37,69.10,67.55(c-6),67.49(c-6’),67.34,67.28,66.14,64.51,57.32(c-2),55.78(c-2’),46.00,38.78, 38.64,33.50,33.41,33.13,30.89,28.67,28.63,28.61,28.59,28.54,28.51,28.31,28.17,24.54,24.15,24.07, 24.03,21.66,18.16,16.82,13.12,13.10,-4.91,-6.31.
[0210]
(2)
[0211]
将化合物6-a(50mg,0.04mmol)与化合物10(51mg,0.049mmol,1.2eq)投入反应瓶,氮气保护加入1ml超干二氯甲烷,-20℃加入稀释100倍的tfoh(0.25mg,0.00167mmol,0.05eq)。1h后化合物6-a(给体)消耗完,化合物10(受体)未消耗完。加甲醇淬灭三乙胺调为中性。柱色谱纯化 (甲苯:乙酸乙酯=12:1)得到化合物11-a(49mg,58.5%)。由于产物11-a与受体10极性接近,与给体6-a极性相差较大。因此,当受体10过量时,虽然反应转化率基本相当,但分离收率较低。
[0212]
(3)
[0213]
将化合物6-a(100mg,0.081mmol,1.2eq)与化合物10(69mg,0.067mmol)投入反应瓶,氮气保护加入1ml超干二氯甲烷,-20℃加入稀释100倍的tfoh(0.5mg,0.0033mmol,0.05eq)。1h 后给体受体均未消耗完,延长反应时间无变化。加甲醇淬灭三乙胺调为中性。柱色谱纯化(甲苯:乙酸乙酯=12:1)得到化合物11-a(58mg,0.0278mmol,41.7%)。
[0214]
(4)
[0215]
将化合物6-a(200mg,0.162mmol,1.5eq)与化合物10(110mg,0.107mmol)投入反应瓶,氮气保护加入1ml超干二氯甲烷,-20℃加入稀释100倍的tfoh(0.8mg,0.0053mmol,0.05eq)。1h 后给体受体均未消耗完,延长反应时间无变化。加甲醇淬灭三乙胺调为中性。柱色谱纯化(甲苯:乙酸乙酯=12:1)得到化合物11-a(107mg,48.2%)。
[0216]
(5)
[0217]
将化合物6-a(200mg,0.162mmol,1.5eq)与化合物10(110mg,0.107mmol)投入反应瓶,氮气保护加入1ml超干二氯甲烷,-20℃加入稀释100倍的tfoh(2.43mg,0.0162mmol,0.15eq)。1h 后给体消耗完,。加甲醇淬灭三乙胺调为中性。柱色谱纯化(甲苯:乙酸乙酯=12:1)得到化合物11
-ꢀ
a(192mg,86.48%)。tof-ms:m/z:2088.14[m na]
。
[0218]
实施例13化合物12-a的制备
[0219][0220]
在反应瓶中将化合物11-a(860mg,0.029mmol)溶于ch2cl2(10ml),n2条件下加入锌粉(2.7g, 40mmol)和醋酸(2.7ml,45mmol)室温下搅拌2h,反应结束后过滤,滤液用甲苯共沸旋干并柱层析(pe:ea=6:1)得到化合物12-a。tof-ms:m/z 1912.06[m na]
。
[0221]
实施例14化合物13-a(mpl-8)的制备
[0222][0223]
将实施例13中制备得到的化合物12-a溶于超干dcm(10ml),n2置换于-10℃下加入edc
·
hcl (2g,10.43mmol)和脂肪链19-a(1.48g,3.47mmol),反应搅拌12h后旋干柱层析纯化(甲苯/乙酸乙酯=10:1)得到化合物13-a(680mg,71%,无色透明糖浆)。tof-ms:m/z 2323.15[m na]
。
[0224]
实施例15化合物14-a的制备
[0225][0226]
在反应瓶中将化合物13-a(580mg,0.252mmol)溶于dmf(12ml),n2条件下加入三乙胺(12ml, 86mmol),室温搅拌过夜,将反应液旋干经柱层析纯化(pe:ea=6:1)得到化合物14-a。tof-ms:m/z 2030.2[m na]
。
[0227]
实施例16化合物15-a的制备
[0228][0229]
将实施例15中制备得到的化合物14-a溶于超干ch2cl2(5ml),n2置换于室温下加入edc
·
hcl (295mg,1.53mmol)和脂肪链20(296mg,0.77mmol),反应液在室温下搅拌12h,旋干柱层析纯化(甲苯/乙酸乙酯=5:1)得到化合物15-a(480mg,收率78%,无色透明糖浆)。tof-ms:m/z 2468.4[m na]
。
[0230]
实施例17化合物16-a的制备
[0231][0232]
将化合物15-a(300mg,0.123mmol)溶于thf(10ml),
–
40℃下加入溶于吡啶(9ml)的hf /py(3ml,65-70%)溶液。升温至室温并搅拌过夜。反应结束后加入饱和碳酸氢钠水溶液淬灭,加入氯仿萃取多次。将有机层干燥过滤并浓缩,通过c18填料纯化(ch3cn,meoh/ea=8:1,meoh依次洗脱)得到16-a(0.214g,75%,白色固体)。
[0233]
δ
h
(600mhz,cdcl3)7.77
–
7.24(28h,m),6.28(1h,d,j 9.5,nh),6.22(1h,d,j 7.4,nh’),5.77
–ꢀ
5.70(1h,m,allyl),5.67
–
5.59(1h,m,allyl),5.44
–
5.31(3h,m,h-3,h-1’,h-3’),5.22
–
5.03(6h,m, allyl,h-1,lipid-h-3),4.99(1h,t,j 5.8,lipid-h-3),4.67
–
4.57(5h,m,nap),4.52
–
4.42(3h,m,nap), 4.35
–
4.21(5h,m,h-4’,diallyl),4.17(1h,td,h-2),4.05(1h,d,j 7.2,h-5),3.87(1h,d,j 11.7,h-6’), 3.80
–
3.74(3h,m,lipid-h-3*2,h-6’),3.67
–
3.56(4h,m,h-4’,h-6*2,h-5’),3.32
–
3.28(1h,m,h-2’), 3.27(1h,t,j 11.6,7.7,h-4),2.55
–
2.43(3h,m),2.28
–
2.11(9h,m),1.45
–
1.37(8h,m),1.22
–
1.10 (108h,m),0.80(18h,dt,j 7.0,5.1).
[0234]
δ
c
(151mhz,cdcl3)173.28,172.55,170.87,170.29,169.85,168.98,135.06,134.48,133.92,132.31, 132.23,132.19,132.10,131.96,131.90,131.86,131.22,131.17,131.11,131.06,127.15,127.09,127.05, 126.91,126.83,126.66,126.57,125.46,125.41,125.36,125.24,125.06,124.99,124.93,124.88,124.85, 124.82,124.73,124.64,124.61,117.45,117.30,98.02(c-1’),90.43(c-1),75.46(c-4),74.52,73.50,73.23, 73.09,72.86(c-5’),72.51(c-3),71.64(c-3’),70.60(c-5),70.36,69.75,69.33,67.55(c-4’),67.51,67.28(c
-ꢀ6’
),67.24,66.43(c-6),59.37,55.37(c-2’),51.48(c-2),41.01,40.61,38.95,38.86,33.63,33.54,33.40,33.34, 30.92,28.64,28.56,28.35,28.21,24.17,24.06,23.89,21.67,13.09.
[0235]
实施例18化合物17-a的制备
[0236][0237]
(1)
[0238]
将化合物16-a(240mg,0.103mmol),pph3(25mg,0.095mmol)加入反应瓶中,氮气保护后加入thf(10ml),tea(125μl,0.9mmol),hcooh(75μl,1.957mmol),pd(ph3)4(25mg,0.02mmol, 0.2eq)。25℃反应5h未反应完全,继续反应12h后原料消失,ms显示有未脱完的allyl,旋干过c18 柱:ch3cn with 0.1%tea,meoh with 0.1%tea,ch2cl2/meoh with 0.1%tea依次洗脱,得到化合物17-a(184mg,79.65%)。
[0239]
(2)
[0240]
将化合物16-a(240mg,0.103mmol),pph3(50mg,0.190mmol)加入反应瓶中,氮气保护后加入 thf(10ml),tea(250μl,1.8mmol),hcooh(150μl,3.914mmol),pd(ph3)4(50mg,0.043mmol, 0.4eq)。25℃反应1.5h后反应完全,旋干过c18柱:ch3cn with 0.1%tea,meoh with 0.1%tea, ch2cl2/meoh with 0.1%tea得到化合物17-a(225mg,97.4%)。tof-m:m/z:2248.76[m-h]
。
[0241]
由于每分子含有两当量allyl,当pph3及其它试剂达到常规的用量(例如pph3与allyl的摩尔比为 0.9:1-1.1:1左右)时,即可高收率的得到化合物17。
[0242]
实施例19化合物18-a的制备
[0243][0244]
(1)
[0245]
将化合物17-a(50mg,0.022mmol)与ddq(1.1g,4.84mmol,220eq)加入反应瓶中,氮气保护下加入干燥chcl3(5ml)。30℃超声60min后结束反应。加入一滴三乙胺淬灭,将反应液旋干。向旋干的反应瓶中加入乙腈,倒入c18填料的柱色谱中脱色纯化。待过量ddq完全除去,ch2cl2:meoh =3:1得到终产品化合物18-a(25mg,67.6%)。(由于ddq大大超出了常规用量,在投料规模较小的情况下,虽然反应收率可达理论值,但是大量残留的ddq影响了后处理纯化的收率)
[0246]
(2)
[0247]
将化合物17-a(50mg,0.022mmol)与ddq(10mg,0.044mmol,2eq)加入反应瓶中,氮气保护下加入干燥chcl3(5ml)。30℃超声60min后结束反应。加入一滴三乙胺淬灭,将反应液旋干。向旋干的反应瓶中加入乙腈,倒入c18填料的柱色谱中脱色纯化。待过量ddq完全除去,ch2cl2:meoh =3:1得到终产品化合物18-a(17mg,45.9%)。如(4),通过延长反应时间,可大大提高收率。
[0248]
(3)
[0249]
将化合物17-a(50mg,0.022mmol)与ddq(40mg,0.177mmol,8eq)加入反应瓶中,氮气保护下加入干燥chcl3(5ml)。30℃超声60min后反应完全。加入一滴三乙胺淬灭,将反应液旋干。向旋干的反应瓶中加入乙腈,倒入c18填料的柱色谱中脱色纯化。待过量ddq完全除去,ch2cl2:meoh =3:1得到终产品化合物18-a(29mg,78.4%)。如(4),通过延长反应时间,
可大大提高收率。
[0250]
(4)
[0251]
将化合物17-a(50mg,0.022mmol)与ddq(40mg,0.177mmol,8eq)加入反应瓶中,氮气保护下加入干燥chcl3(5ml)。30℃超声80min后反应完全。加入一滴三乙胺淬灭,将反应液旋干。向旋干的反应瓶中加入乙腈,倒入c18填料的柱色谱中脱色纯化。待过量ddq完全除去,ch2cl2:meoh =3:1得到终产品化合物18-a(35.8mg,95.26%,hplc纯度:98.5%)。tof-ms:m/z:1689.36[m-h]
。
[0252]
化合物18-b系列实施例
[0253]
实施例20化合物13-b的制备
[0254][0255]
在反应瓶中将11-a(400mg,0.194mmol)溶于ch2cl2(5ml),n2条件下加入锌粉1.2g)和醋酸(1.2 ml)室温下搅拌2h,反应结束后过滤,滤液用甲苯共沸旋干并柱层析(pe:ea=6:1)得到化合物12-a(经 ms验证tof-ms:m/z 1915.49[m na]
)
[0256]
将化合物12-a溶于超干dcm(5ml),n2置换于-10℃下加入edc
·
hcl(0.892g,4.656mmol)和脂肪链19-c(749mg,1.552mmol)(cas:93390-37-5),反应搅拌12h后旋干柱层析纯化(甲苯/乙酸乙酯=10:1)得到化合物13-b(722.7mg,63.4%,无色透明糖浆)。tof-ms:m/z 2380.26[m na]
。
[0257]
实施例21化合物14-b的制备
[0258][0259]
在反应瓶中将化合物13-b(600mg,0.254mmol)溶于dmf(12ml),n2条件下加入三乙胺(12ml),室温搅拌过夜,将反应液旋干经柱层析纯化(pe:ea=6:1)得到化合物14-b(经ms验证)。tof-ms:m/z 2158.02[m na]
。
[0260]
实施例22化合物15-b的制备
0.1%tea,ch2cl2/meoh with 0.1%tea得到化合物17-b(428mg,94.3%)。tof-m:m/z:2330.16[m na]
。
[0269]
实施例25化合物18-b的制备
[0270][0271]
将化合物17-b(90mg,0.039mmol)与ddq(71mg,0.312mmol,8eq)加入反应瓶中,氮气保护下加入干燥chcl3(10ml)。30℃超声80min后反应完全。加入一滴三乙胺淬灭,将反应液旋干。向旋干的反应瓶中加入乙腈,倒入c18填料的柱色谱中脱色纯化。待过量ddq完全除去, ch2cl2:meoh=3:1得到终产品化合物18-b(65mg,95.26%)。tof-ms:m/z:1745.47[m-h]
。hplc纯度:97.5%。
[0272]
化合物18-c系列实施例
[0273]
实施例26化合物2-c的制备
[0274][0275]
将化合物19-b(cas:163310-36-9)(2.613g,5.93mmol)与edc
·
hcl(1.136g,5.93mmol,1.2eq)溶于ch2cl2(25ml),室温搅拌15min后加入化合物1(3g,4.94mmol)和dmap(0.03g,0.247mmol, 0.05eq),室温下搅拌10h反应完,反应液依次用ch2cl2(50ml)和饱和nahco3(30ml)洗涤。分液干燥浓缩后经硅胶柱色谱纯化(石油醚/乙酸乙酯=10:1)得到化合物2-c(4.29g,84.3%,无色糖浆)。 tof-ms:m/z:1052.7[m na]
。
[0276]
实施例27化合物3-c的制备
[0277][0278]
将化合物2-c(1g,0.97mmol)溶与thf(10ml),依次在冰浴条件下加入bh3·
me3n(0.28g,3.783 mmol,3.9eq),alcl3(0.76g,5.7mmol),h2o(35mg,1.92mmol,1.9eq)反应液在室温下搅拌1.5h。加水(10ml)、1m hcl溶液(10ml)淬灭,反应液用ch2cl2(15ml)分液,干燥浓缩后经硅胶柱色谱纯化(石油醚/乙酸乙酯=10:1)得到化合物3-c(0.882g,87.6%,无色油状液体)。tof
-ꢀ
ms:m/z:1059.42[m na]
.
[0279]
实施例28化合物4-c的制备
[0280][0281]
将化合物3-c(0.5g,0.482mmol)与四氮唑(102mg,1.45mmol,3eq)溶于超干乙腈(10ml),加入烯丙基配体(己二烯-n,n-二异丙基亚磷酰胺,0.236g,0.964mmol,2eq),室温下反应40min后原料消耗完。-40℃下将mcpba(207.9mg,1.205mmol,2.5eq)溶于超干二氯甲烷(15ml),加入上述反应体系,缓慢升至-10℃,40min反应完加饱和硫代硫酸钠溶液(20ml)淬灭,饱和碳酸氢钠洗涤 (40ml),干燥旋干硅胶柱色谱纯化(石油醚:乙酸乙酯=8:1)得化合物4-c(0.537g,93.3%)。tof
-ꢀ
ms:m/z:1214.8[m na]
.
[0282]
实施例29化合物5-c的制备
[0283][0284]
将化合物4-c(1.5g,1.26mmol)溶于thf(45ml),
–
40℃加入稀释于30ml吡啶的hf/pyridine(4.8 ml,65
–
70%)。缓慢升至室温反应12h,反应液用nahco3(80ml)淬灭,加入ch2cl2(100ml)分液,干燥旋干硅胶柱色谱纯化(石油醚:乙酸乙酯=4:1)得化合物5-c(1.26g,93.4%,白色固体)。tof
-ꢀ
ms:m/z:1100.53[m na]
。
[0285]
实施例30化合物6-c的制备
[0286][0287]
将化合物5-c(1.3g,1.21mmol)溶于超干ch2cl2(20ml),n2保护下加入2,2,2-三氟-n-苯乙酰亚胺氯(1.50g,7.24mmol,6eq)与dbu(368mg,2.42mmol,2eq),室温反应30mins。将反应液浓缩柱色谱纯化(石油醚:乙酸乙酯=15:1with 0.1%et3n)得到化合物6-c(1.19g,79%,无色糖浆)。tof
-ꢀ
ms:m/z:1271.65[m na]
。
[0288]
实施例31化合物11-c的制备
[0289][0290]
在反应瓶中加入受体化合物10(700mg,0.684mmol),给体化合物6-c(1.28g,1.026mmol,1.5eq) 分子筛(600mg),n2条件下加入超干ch2cl2(10ml),在-20℃下加入二氯甲烷稀释100倍的 tfoh(20μl,0.137mmol,0.2eq),
–
20℃下搅拌40min;加入甲醇(5ml)淬
灭,三乙胺调ph至8。反应液用ch2cl2(35ml)和饱和nahco3(10ml)洗涤。有机相干燥浓缩,经柱层析纯化(pe:ea=10:1) 得到糖苷化产物11-c(1.27g,89%)。tof-ms:m/z 2104.9[m na]
。
[0291]
实施例32化合物12-c的制备
[0292][0293]
在反应瓶中将11-c(500mg,0.24mmol)溶于ch2cl2(5ml),n2条件下加入锌粉1.5g)和醋酸 (15ml)室温下搅拌2h,反应结束后过滤,滤液用甲苯共沸旋干并柱层析(pe:ea=6:1)得到化合物 12-c(经ms验证tof-ms:m/z 1929.5[m na]
)。
[0294]
实施例33化合物13-c的制备
[0295][0296]
将化合物12-c溶于超干dcm(5ml),n2置换于-10℃下加入edc
·
hcl(1.1g,5,76mmol)和脂肪链19-a(819mg,1.92mmol),反应搅拌12h后旋干柱层析纯化(甲苯/乙酸乙酯=10:1)得到化合物13
-ꢀ
c(441mg,79.4%,无色透明糖浆)。tof-ms:m/z 2338.23[m na]
。
[0297]
实施例34化合物14-c的制备
[0298][0299]
在反应瓶中将化合物13-c(600mg,0.259mmol)溶于dmf(12ml),n2条件下加入三乙胺(12ml),室温搅拌过夜,将反应液旋干经柱层析纯化(pe:ea=6:1)得到化合物14-c(经ms验证)。tof-ms:m/z 2115.9[m na]
。
[0300]
实施例35化合物15-c的制备
[0301][0302]
将化合物14-c溶于超干ch2cl2(5ml),n2置换于室温下加入edc
·
hcl(397.2mg,2.072mmol)和脂肪链20(398.4mg,1.036mmol),反应液在室温下搅拌12h,旋干柱层析纯化(甲苯/乙酸乙酯=5:1) 得到化合物15-c(518.8mg,81.4%,无色透明糖浆)。tof-ms:m/z 2482.47[m na]
。
[0303]
实施例36化合物16-c的制备
[0304][0305]
将化合物15-c(400mg,0.162mmol)溶于thf(18ml),
–
40℃下加入溶于吡啶(12ml)的hf /py(4ml,65-70%)溶液。升温至室温并搅拌过夜。反应结束后加入饱和碳酸氢钠水溶液淬灭,加入氯仿萃取多次。将有机层干燥过滤并浓缩,通过c18填料纯化(ch3cn,meoh/ea=8:1,meoh) 得到16-c(0.293g,76.9%,白色固体)。tof-ms:m/z 2368.26[m na]
。
[0306]
实施例37化合物17-c的制备
[0307][0308]
将化合物16-c(459mg,0.196mmol),pph3(90mg,0.370mmol)加入反应瓶中,氮气保护后加入 thf(20ml),tea(490μl,3.5mmol),hcooh(290μl,7.8mmol),pd(ph3)4(90mg,0.08mmol)。25℃反应1.5h后反应完全,旋干过c18柱:ch3cn with 0.1%tea,meoh with 0.1%tea,ch2cl2/meohwith 0.1%tea得到化合物17-c(405mg,91.5%)。tof-ms:m/z 2288.05[m na]
。
[0309]
实施例38化合物18-c的制备
[0310][0311]
将化合物17-c(100mg,0.044mmol)与ddq(80mg,0.353mmol,8eq)加入反应瓶中,氮气保护下加入干燥chcl3(10ml)。30℃超声80min后反应完全。加入一滴三乙胺淬灭,将反应液旋干。向旋干的反应瓶中加入乙腈,倒入c18填料的柱色谱中脱色纯化。待过量ddq完全除去, ch2cl2:meoh=3:1得到终产品化合物18-c(68mg,91.5%)。tof-ms:m/z:1703.4[m-h]
。hplc纯度:96.5%
[0312]
对比实施例1
[0313]
将化合物17(50mg,0.022mmol)与pd(400mg)加入氢化釜中,溶于thf:h2o=4:1(20ml), 1mpa,30℃反应24h。加入一滴三乙胺淬灭,将反应液过滤旋干。使用c18填料的柱色谱中脱色纯化得到终产品化合物18(12mg,32.4%)。通过tlc板层显示,原料有剩余,杂质点多,延长时间原料未减少。收率和纯度明显差于上述实施例。
[0314]
由上可见,(1)本发明使用烯丙基配体在后续避免了氢化反应,用四三苯基膦的条件1.5h即可反应完。同样使用c18填料柱简单脱色后即可得到终产品。避免了现有技术中采用氢化脱苄基保护基带来的杂质多,收率较低,且纯化方式复杂的问题。具有明显更优的效果。
[0315]
(2)与现有技术需要使用钯碳氢化反应20h以上,再反复过滤过离子柱色谱,收率较低(仅能达到50%左右)相比较,本发明使用nap保护基的方法简化了操作,优化后,脱保护步骤收率可达到 91.5%以上,纯度达到97%(测定方法参照:hplc-elsd法测定blp25脂质体疫苗中mpl的含量,中国药事2012年第26卷第5期,王明娟,王琰,胡昌勤)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

