通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法及应用
技术领域
1.本发明属于生物技术领域,具体涉及一种通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法及应用。
背景技术:
2.近年来随着全球气候变暖的加剧,全国大部分香菇主产区尤其是湖北、浙江等地暴发了受到木霉菌感染的菌棒腐烂病,对香菇产业的健康发展造成了极大的危害。尤其是香菇菌丝越夏及转色阶段,更容易受到木霉菌的侵染,受到高温的影响是导致香菇菌丝对杂菌抗性能力降低的最主要原因之一。因此,鉴定香菇对木霉的抗性,筛选出高抗香菇菌种,对于香菇的生产是非常重要的。
3.然而,现有技术关于香菇对木霉抗性的研究只处于表观描述阶段,没有任何的有效的抗性测定和验证方法,妨碍了香菇对木霉抗性的定性定量研究,阻碍了优秀品种的选育。
技术实现要素:
4.为了解决上述技术问题,本发明提供了一种通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法及应用。
5.本发明的目的是提供一种通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,包括以下步骤:
6.利用香菇dna基因和载体pcambia1300
‑
g构建letlp1过表达载体 pcambia1300
‑
oe
‑
letlp1;
7.将pcambia1300
‑
oe
‑
letlp1转化入农杆菌中,挑取单个阳性克隆备用;
8.转化:香菇菌株菌丝在培养基上活化后,浸入活化好的含 pcambia1300
‑
oe
‑
letlp1农杆菌菌液;将香菇菌株菌丝接种至含有as的共培养固体培养基co
‑
im上,共培养;之后,将菌丝块从共培养培养基挑取下来,用灭菌的蒸馏水清洗菌块,浸泡在含头孢噻肟霉素的无菌水中;之后将菌丝块吸干表面水分,转移至含有潮霉素和头孢噻肟霉素的myg固体培养基上,25℃培养7
‑
12d;
9.鉴定和筛选:挑取有潮霉素抗性的菌株进行鉴定,筛选出的阳性菌株为能超表达香菇letlp1基因的菌株,该菌株对深绿木霉的抗性提高。
10.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,构建letlp1过表达载体具体包括如下步骤:
11.第一步:以香菇的dna为模板,以o
‑
letlp1
‑
f和o
‑
letlp1
‑
r为引物,对letlp1基因全长片段进行扩增;以香菇的dna为模板,以o
‑
legpd promotor
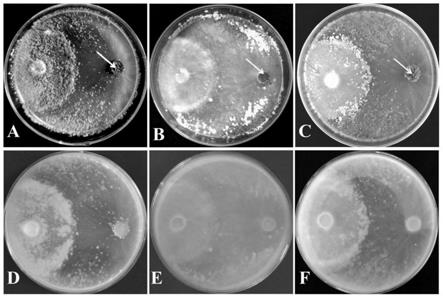
‑
f和o
‑
legpd promotor
‑
r为引物,对香菇legpd启动子片段进行扩增;
12.o
‑
letlp1
‑
f核苷酸序列如下:
13.ccacctcaaacttcggaattctcaggggcaaaatgtaacagcata;
14.o
‑
letlp1
‑
r核苷酸序列如下:
15.ccattgactgcttgaatatgatgaagaattctatcattatctctgc;
16.o
‑
legpd promotor
‑
f核苷酸序列如下:
17.catattcaagcagtcaatggattgga;
18.o
‑
legpd promotor
‑
r核苷酸序列如下:
19.tctagaggatccccgggtacccgaagtttgaggtggttgcg;
20.第二步:电泳检测并回收legpd启动子片段和letlp1基因全长片段;
21.第三步:用限制性内切酶kpn
‑
i和ecor
‑
i对载体pcambia1300
‑
g进行双酶切,并将线性化载体的pcambia1300
‑
g、legpd启动子片段和letlp1基因全长片段通过重组酶连接。
22.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,第一步中,
23.扩增的反应体系如下:2
×
phanta max buffer 20μl,引物各1μl,dntp mix 1μl,phanta max super
‑
fidelity dna polymerase 1μl,模板量为200ng,补ddh2o 至40μl体系;
24.上述扩增的反应条件:95℃预变性3min,95℃变性15s,60℃退火15s, 72℃延伸1min,循环数34,72℃延伸5min。
25.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,第三步中,
26.重组酶连接体系为:酶切线性化片段pcambia1300
‑
g 160ng,legpd启动子20ng,letlp1全长基因片段30ng,2
×
basic assembly mix 5μl,补ddh2o 至10μl。
27.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,所述农杆菌菌株为eha105。
28.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,所述香菇菌株为武香一号、y55或y3334。
29.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,所述转化的具体步骤为:香菇菌株在myg固体培养基上活化6
‑
8d后,取直径 5mm的菌丝块,浸入活化好的含pcambia1300
‑
oe
‑
letlp1农杆菌菌液20
‑
30 min,每隔5
‑
10min摇匀一次;之后,将菌丝块接种至含有200μmol/l as的共培养固体培养基co
‑
im上,25
±
1℃共培养3
‑
4d;之后,将菌丝块从共培养培养基挑取下来,用灭菌的蒸馏水清洗,浸泡在含400μg/ml头孢噻肟霉素的无菌水中20
‑
30min,每隔5
‑
10min摇匀一次;菌丝块吸干表面水分,转移至含有潮霉素和300μg/ml头孢噻肟霉素的myg固体培养基上,25
±
1℃培养7
‑
12d。
30.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,对于y55菌株,潮霉素终浓度为70μg/ml;对于y3334菌株,潮霉素终浓度为9μg/ml。
31.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,所述鉴定的方法包括pcr或者qrt
‑
pcr方法。
32.优选的,上述通过超表达香菇tlp基因提高香菇对深绿木霉抗性的方法,将letlp1过表达载体pcambia1300
‑
oe
‑
letlp1用于增强了香菇菌株对深绿木霉的抗性。
33.与现有技术相比,本发明具有以下有益效果:
34.1、本发明采用致病力强的深绿木霉(trichoderma atrovide)研究了香菇与木霉之间的互作反应。通过平板对峙试验研究了与210个香菇菌株对深绿木霉的抗性反应,初步建立了香菇对木霉的抗性鉴定技术。应用该技术详细对56个香菇核心种质资源菌株(包括21份栽培香菇菌株和35份野生香菇菌株)的木霉抗性进行了详细的鉴定与分级,综合评价筛选出多株强抗和唯一的一株弱抗香菇菌株。
35.2、本发明选择抗性显著差异的两个香菇菌株y3334和y55(此两种菌记载于“江晴.香菇种质资源群体抗木霉能力分化的初步研究[d].华中农业大学, 2017”中),其中y3334是对深绿木霉抗性较强的菌株,y55是对深绿木霉完全不抗的菌株,通过扫描电镜分析了y3334、y55与深绿木霉不同对峙时间的菌丝形态,结果发现在平板和代料两种基质中,弱抗菌株y55的菌丝体都易被深绿木霉的孢子重寄生。
[0036]
具体的,本发明利用对深绿木霉抗性有显著差异的两个香菇菌株(y3334、 y55)分别与同一株深绿木霉进行对峙培养,取对峙后24小时的香菇菌丝提取总rna进行了链特异性转录组测序,测序平台illumina hiseq 4000,双末端各测100bp,每个样品测3次重复,每个样品测4g数据量。测序结果表明,利用 fastqc质控软件得到的clean data能有效匹配到香菇参考基因组的比例在 50%
‑
70%之间。利用cufflinks软件计算得知,对深绿木霉强抗的香菇菌株有4550 个差异表达基因,对深绿木霉弱抗的香菇菌株有761个差异表达基因。进一步将强抗香菇菌株的差异表达基因与弱抗菌株的差异表达基因进行比较,筛选出在强抗香菇菌株中显著上调表达而在弱抗香菇菌株中为下调表达的基因一共 377个,对这377个目标基因进行功能注释,发现有与病程相关、营养利用,分泌蛋白、膜融合蛋白、转录因子等相关基因,对其中与抗病相关的基因tlp (letlp1基因)进行基因全长序列的克隆和分析,并将强抗菌株y3334的 letlp1基因中。
[0037]
3、本发明以强抗菌株y3334 cdna为模板扩增letlp1基因全长片。将线性化载体pcambia1300
‑
g、legpd启动子和letlp1基因全长片段通过重组酶连接,构建y55菌株letlp1基因过表达载体,获得超表达转化子,研究了 letlp1基因过表达对y55菌株深绿木霉抗性的影响,结果发现超表达letlp1 基因能加强弱抗香菇菌株对深绿木霉的抗性,因此可以通过超表达letlp1基因的方法加强香菇菌株对木霉的抗性。
附图说明
[0038]
图1为本发明实施例1构建的重组过表达载体pcambia1300
‑
oe
‑
letlp1 图谱及其鉴定结果;
[0039]
其中,a为pcambia1300
‑
oe
‑
letlp1,b为大肠杆菌单菌落pcr扩增鉴定结果,1
‑
5泳道为大肠杆菌菌落,m:1kb dna ladder;c为单酶切验证结果, 1
‑
3泳道为酶切鉴定结果,m:1kb dna ladder;d为农杆菌菌落pcr鉴定结果, 1
‑
2泳道为农杆菌菌落;m:1kb dna ladder,p为pcambia1300
‑
oe
‑
letlp1;
[0040]
图2为本发明实施例2的y55菌株letlp1基因过表达转化子验证结果;
[0041]
a:letlp1基因过表达转化子pcr鉴定;1
‑
10泳道分别表示编号 letlp1
‑
oe
‑
1到letlp1
‑
oe
‑
10,wt泳道为y55,p泳道为 pcambia1300
‑
oe
‑
letlp1,m:bm2000 dna marker;
[0042]
b:letlp1
‑
oe
‑
1到letlp1
‑
oe
‑
10的letlp1基因荧光定量pcr分析;1
‑
10 分别对应编号letlp1
‑
oe
‑
1到letlp1
‑
oe
‑
10的letlp1基因表达量,wt表示 y55中letlp1基因的原有
表达量;*p<0.05**p<0.01。
[0043]
图3为本发明实施例2的letlp1过表达对y55深绿木霉抗性影响图;
[0044]
a和d:深绿木霉孢子覆盖y55菌丝平板正面和反面图;b和e:letlp1
‑
oe
‑
3 菌丝表面有少量的深绿木霉孢子平板正面和反面图;c和f:letlp1
‑
oe
‑
10菌丝表面有少量的深绿木霉孢子平板正面和反面图;
[0045]
其中,白色箭头表示香菇接种块;灰色箭头表示深绿木霉接种块。
具体实施方式
[0046]
为了使本领域技术人员更好地理解本发明的技术方案能予以实施,下面结合具体实施例和附图对本发明作进一步说明。
[0047]
在本发明的描述中,如未特殊说明,所用试剂均为市售,所用方法均为本领域常规技术。本发明所用引物信息参见表1.
[0048]
表1引物信息
[0049][0050]
注:对于表1中最后两个同源重组引物,小写字母表示同源臂,大写字母为基因片段的引物。
[0051]
实施例1 letlp1过表达载体的构建
[0052]
(1)letlp1基因克隆
[0053]
用primer5进行letlp1基因的开放阅读框(open reading frame,orf) 引物设计letlp1
‑
f和letlp1
‑
r(表1)。以y3334的dna和cdna为模板,扩增letlp1基因。扩增反应体系如下(40μl):2
×
phanta max buffer 20μl,引物(10μm)各1μl,dntp mix 1μl,phanta max super
‑
fidelity dna polymerase1μl,模板量为200ng,补ddh2o至40μl体系。反应条件:95℃预变性3min,95℃变性15s,60℃退火15s,72℃延伸1min,循环数34,72℃延伸5min。用1%(w/v)琼脂糖凝胶电泳检测并切胶回收目的条带,用sanprep柱式dna 回收试剂盒回收letlp1基因片段。将回收的目的条带利用pclone007 bluntsimple vector kit试剂盒连接至克隆载体pcambia1300
‑
g上,并转化至大肠杆菌trans1
‑
t1感受态细胞中。挑取单菌落进行pcr验证,并送至武汉天一华煜基因科技有限公司进行测序。
[0054]
(2)重组过表达载体的构建
[0055]
通过ce design(http://www.vazyme.com)进行构建载体所用引物的设计(表 1),酶切位点选用ecor
‑ⅰ
和kpn
‑ⅰ
,通过同源重组方法构建重组过表达载体。
[0056]
具体方法如下:
[0057]
(2.1)片段扩增和回收
[0058]
以香菇wx1(武香一号)的dna为模板,以o
‑
letlp1
‑
f和o
‑
letlp1
‑
r 为引物,对letlp1基因全长片段进行扩增;以香菇wx1的dna为模板,以 o
‑
legpd promotor
‑
f和o
‑
legpd promotor
‑
r为引物,对香菇legpd启动子片段进行扩增;以香菇y3334 cdna为模板。
[0059]
上述扩增的反应体系如下(40μl):2
×
phanta max buffer 20μl,引物(10μm) 各1μl,dntp mix 1μl,phanta max super
‑
fidelity dna polymerase 1μl,模板量为200ng,补ddh2o至40μl体系。
[0060]
上述扩增的反应条件:95℃预变性3min,95℃变性15s,60℃退火15s, 72℃延伸1min,循环数34,72℃延伸5min。
[0061]
用1%(w/v)琼脂糖凝胶电泳检测并切胶回收目的条带,用sanprep柱式 dna回收试剂盒回收legpd启动子片段和letlp1基因全长片段。
[0062]
(2.2)线性化载体的酶切回收
[0063]
首先将含有骨架载体pcambia1300
‑
g的大肠杆菌在37℃,200r/min过夜培养,提取质粒。用限制性内切酶kpn
‑
i和ecor
‑
i(thermo scientific)对质粒pcambia1300
‑
g进行双酶切,37℃酶切1h后,用1%(w/v)琼脂糖凝胶电泳检测并切胶回收目的条带,用san prep柱式dna回收试剂盒回收线性化质粒。酶切体系:pcambia1300
‑
g质粒30μg,kpn
‑
i和ecor
‑
i各2μl,10
×
fastdigestbuffer 4μl,补ddh2o至40μl。需要说明的是,pcambia1300
‑
g质粒是原35s 启动子被香菇legpd启动子替换,以启动潮霉素b磷酸转移酶基因表达该载体由华中农业大学应用真菌研究所保存,是一个经过改造的载体,具有唯一性(yanl,xu r,zhou y,gong yu,dai sheng,liu hai,bian yin.effects of mediumcomposition and genetic background on agrobacterium
‑
mediated transformationefficiency of lentinula edodes.genes,2019,10(6):467)。
[0064]
(2.3)重组构建过表达载体
[0065]
将线性化载体pcambia1300
‑
g(含letlp1基因保守结构域片段)、legpd 启动子片段和letlp1基因全长片段通过重组酶连接。重组酶连接体系:酶切线性化片段pcambia1300
‑
g 160ng,legpd启动子20ng,letlp1基因片段30ng, 2
×
basic assembly mix 5μl,补ddh2o至10μl。
[0066]
(2.4)转化大肠杆菌和重组质粒验证
[0067]
将连接产物转化至大肠杆菌trans1
‑
t1感受态细胞中,涂布lb平板(含50 μg/ml卡那霉素)。挑取单菌落,用o
‑
letlp1
‑
f和o
‑
legpd promotor
‑
r引物验证重组过表达载体;挑取阳性单菌落送去武汉天一华煜基因科技有限公司进行测序,并提取质粒用xho
‑
i单酶切验证。酶切体系:质粒1μg,xho
‑
i 1μl, 10
×
fastdigest buffer 1μl,ddh2o 10μl。37℃酶切1h后,用1%(w/v)琼脂糖凝胶电泳检测。重组过表达载体被命名pcambia1300
‑
oe
‑
letlp1。
[0068]
上述获得的验证正确的重组过表达载体pcambia1300
‑
oe
‑
letlp1转化至农杆菌eha105中。
[0069]
重组过表达载体pcambia1300
‑
oe
‑
letlp1参见图1a,其转入大肠杆菌(e. coil)感受态细胞trans1
‑
t1后,挑取5个单菌落进行pcr扩增(legpd和letlp1 基因全长)验证,其中
4个单菌落有1785bp的条带(图1b)。挑取三个阳性单菌落于lb(50μg/ml kan
)液体培养基中进行扩大培养并提取质粒,采用限制性内切酶xho i单酶切,结果显示有7.6kb、1.8kb和1.3kb的3条带(图1c)。阳性单菌落测序分析显示y3334菌株的letlp1编码序列与基因组测序单核菌株w1
‑
26(该菌株记载于“颜莲莲,徐瑞平,龚钰华,et al.香菇遗传背景差异和培养基营养成分影响农杆菌介导的遗传转化效率[c]//多彩菌物美丽中国——中国菌物学会2019年学术年会”中)相似度为97%,表明构建的载体 pcambia1300
‑
oe
‑
letlp1是正确的。将阳性质粒pcambia1300
‑
oe
‑
letlp1 转化入农杆菌菌株eha105中,挑取单个阳性克隆保存于
‑
80℃备用(图1d)。
[0070]
实施例2 tlp基因提高香菇对深绿木霉抗性的验证
[0071]
(1)农杆菌介导的香菇菌丝遗传转化
[0072]
将含有pcambia1300
‑
oe
‑
letlp1的阳性eha105农杆菌菌液划线培养3d,挑取单菌落于1ml lb(50μg/ml kan
)液体培养基,28℃200r/min过夜培养。而后将1ml培养好的农杆菌菌液加入到100ml mm液体基本培养基(50 μg/ml kan
)中,28℃200r/min培养至od600约为0.8。5000r/min离心10min 收集菌体,用适量im诱导培养基重悬菌体至od600约为0.4,添加乙酰丁香酮 (as)至终浓度为200μmol/l,28℃200r/min培养6h左右至od600约0.8。
[0073]
香菇菌株y55和y3334菌丝在myg固体培养基(培养基配方:每升含蛋白胨5.0g,麦芽浸粉3.0g,酵母浸粉3.0g,葡萄糖10.0g,琼脂20.0g,ph6.2
±ꢀ
0.2(25℃),溶剂为蒸馏水)上活化7d后,用灭菌的蓝枪头打取直径5mm的菌丝块,浸入活化好的含pcambia1300
‑
oe
‑
letlp1农杆菌菌液20min,每隔5min 摇匀一次。之后,将菌丝块接种至含有200μmol/l as的共培养固体培养基co
‑
im (添加了as至终浓度为200μmol/l的im培养基)上,25℃共培养3天。之后,将菌丝块从共培养培养基挑取下来,用灭菌的蒸馏水清洗菌块3次,浸泡在含 400μg/ml头孢噻肟霉素的无菌水中20min,每隔5min摇匀一次。菌丝块再用灭菌滤纸稍微吸干表面水分,转移至含有潮霉素(y55 70μg/ml;y3334 9μg/ml) 和300μg/ml头孢噻肟霉素的myg固体培养基上,25℃培养10d左右。
[0074]
(2)转化子筛选和验证
[0075]
(2.1)菌丝研磨和筛选
[0076]
菌丝萌发后,挑取新鲜菌块于灭菌研钵中,加适量无菌水研磨成糊状,均匀涂抹于含潮霉素(y55 70μg/ml;y3334 9μg/ml)的myg固体培养基上,25℃正置培养7
‑
15d。挑取萌发的单菌落在含有潮霉素(y55 70μg/ml;y3334 9μg/ml) myg固体培养基上进行4次筛选的),挑取有潮霉素抗性的菌株进行下一步试验。
[0077]
(2.2)转化子pcr检测
[0078]
挑取能够正常生长的香菇转化子,在含有9μg/ml潮霉素贴有玻璃纸的 myg平板上25℃避光培养10d后采用ctab法提取转化子总dna。
[0079]
dna具体提取步骤如下:
[0080]
1)适量香菇转化子和野生型香菇的菌丝用液氮冷冻并研磨成粉末状,加入 500μl抽提缓冲液(475μl ctab缓冲液:100mmol/l tris
‑
hcl(ph=7.8), 20mmol/l edta,1.4mol/l naac,2%(w/v)ctab;25μl 5%sds);
[0081]
2)充分混匀30s,加入500μl pci(苯酚:氯仿:异戊醇=25:24:1);充分混匀30s,13000r/min,离心10min;上清转移到新的1.5ml离心管中,加入2/3体积的异丙醇,
‑
20℃静
置10min;
[0082]
3)13000r/min,离心2
‑
4min,70%(v/v)酒精洗涤沉淀2次;
[0083]
4)超净工作台吹干酒精,加入50μl ddh2o溶解,
‑
20℃保存备用。
[0084]
用超微量紫外分光光度计测定浓度。以上述转化子的dna为模板进行pcr 扩增,检测转化子是否插入了目的载体。以抗性转化子dna和野生型菌株y55 y3334 dna为模板,以yz
‑
gpd
‑
f和yz
‑
hyg
‑
r(表1)验证y55 letlp1基因过表达转化子。pcr扩增程序:95℃预变性5min,95℃变性30s,58℃退火30s, 72℃延伸1min 20s,34个循环,在72℃最终延伸时间10min。
[0085]
本实施例经过5轮myg培养基(70μg/ml潮霉素)上筛选,获得10个能够在70μg/ml潮霉素培养基正常生长的转化子。采用ctab法提取野生型和转化子基因组dna,并利用yz
‑
gpd
‑
f和yz
‑
hyg
‑
r引物对提取的dna进行特异性扩增,然后进行1%(w/v)琼脂糖凝胶电泳检测。电泳结果显示10个转化子有一条约1.2kb条带(legpd启动子和潮霉素b磷酸转移酶基因部分序列长度 1230bp)(2a),因此这10个转化子被认为阳性转化子并进行下一步验证。
[0086]
(2.3)转化子qrt
‑
pcr分析
[0087]
将pcr鉴定为阳性的香菇转化子,在铺有玻璃纸的myg平板上25℃避光培养10d后,采用rnaisotm试剂盒(takara,大连)提取rna,然后使用 hiscript ii q rt supermix for qpcr( gdna wiper)试剂盒将rna反转录为 cdna。以反转录后的cdna为模板,对letlp1基因进行分析,leactin为内参基因,y55和y3334的cdna为对照。
[0088]
采用aceqtm qpcr sybr green master mix在cfx connect real
‑
timepcr system(bio
‑
rad)上进行qrt
‑
pcr。反应体系为aceqtm qpcr sybrgreen master mix 5μl,引物q
‑
letlp1
‑
f(10μm)和q
‑
letlp1
‑
r(10μm)各0.25 μl,cdna 3μl,ddh2o 1μl。程序设置为95℃3min,95℃20s,60℃30 s,72℃30s;35cycles;60℃,plate read;溶解曲线的温度设置为60℃到90℃,每0.5℃读取一次数据。基因相对表达量计算方法采用2
‑
δδct
(pfaffl m(2001)anew mathematical model for relative quantification in real
‑
time rt
‑
pcr.nucleicacids research 29:2002
–
2007.)。本次试验所有定量分析均以leactin作为内参,野生型菌株的cdna为对照进行后续数据分析。
[0089]
将pcr鉴定的10个阳性转化子分别命名为letlp1
‑
oe
‑
1、letlp1
‑
oe
‑
2、 letlp1
‑
oe
‑
3、letlp1
‑
oe
‑
4、letlp1
‑
oe
‑
5、letlp1
‑
oe
‑
6、letlp1
‑
oe
‑
7、 letlp1
‑
oe
‑
8、letlp1
‑
oe
‑
9、letlp1
‑
oe
‑
10,对这些阳性转化子进行rna的提取,反转录成cdna,采用荧光定量pcr对它们的letlp1基因的表达量进行分析,并以野生型菌株y55为对照,leactin基因作为内参。定量结果发现:有4个转化子的letlp1基因的表达量是野生菌株该基因表达量的2倍以上,且 letlp1
‑
oe
‑
3和letlp1
‑
oe
‑
10将letlp1的表达量分别上调了约5倍和7倍(图2b)。因此,选取这两个转化子进行下一步的验证。
[0090]
(3)深绿木霉抗性测定
[0091]
将转化子letlp1
‑
oe
‑
3和letlp1
‑
oe
‑
10和野生型香菇菌株y55分别在 pda培养基上活化8d,用1ml蓝色枪头打孔,接种于9cm平皿距边沿1cm 位置,25℃培养7d。之后,在平皿另一半相应的位置接种深绿木霉的菌块,25℃对峙培养。刚接触时,在菌丝尖端划起始线,随后再对峙培养15d,观察深绿木霉和香菇菌丝互作的情况,每个菌株有5个重复,拍照记录。采用相互作用区域菌丝代产物颜色的变化以及计算侵染率(深绿木霉菌丝覆盖香菇菌
丝的长度与香菇接种块到接触线的距离之比),使用image j软件的直线工具计算深绿木霉菌丝覆盖香菇菌丝的长度与香菇接种块到接触线的距离之比,spss软件进行单因素方差分析(α=0.05)。
[0092]
香菇菌丝和深绿木霉菌丝接触时计为0d,再进行15d的对峙培养。采用相互作用区域菌丝代产物颜色的变化以及计算侵染率(深绿木霉菌丝覆盖香菇菌丝的长度与香菇接种块到接触线的距离之比),评价letlp1基因在香菇菌丝与深绿木霉菌丝对峙过程中的影响。
[0093]
结果显示:经过15d的对峙培养后,野生型菌株y55和深绿木霉之间不能够形成拮抗线(图3a和d),且深绿木霉菌丝能够完全覆盖y55菌丝(侵染率约100%),在香菇菌丝上形成大量的深绿木霉孢子。对于2个letlp1过表达转化子,过表达转化子letlp1
‑
oe
‑
3的菌丝能够不被深绿木霉菌丝完全覆盖,且对深绿木霉孢子的萌发少于野生型菌株y55(图3b和e);过表达转化子 letlp1
‑
oe
‑
10对深绿木霉的抗性最明显(侵染率约10%),并且阻止深绿木霉菌丝进一步的侵染(图3c和f)。木霉对峙的结果可以发现过表达转化子 letlp1
‑
oe
‑
3(侵染率约40%)对深绿木霉的抗性弱于letlp1
‑
oe
‑
10。
[0094]
上述结果表明,letlp1过表达增强了香菇菌株y55对深绿木霉的抗性,且对深绿木霉的抗性与基因表达的水平呈正相关。
[0095]
需要说明的是,本发明中涉及数值范围时,应理解为每个数值范围的两个端点以及两个端点之间任何一个数值均可选用,由于采用的步骤方法与实施例相同,为了防止赘述,本发明描述了优选的实施例。尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例做出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
[0096]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

