1.本发明涉及甲基丙烯酸酯类共聚物材料及其合成方法和医学材料领域,尤其涉及基于光催化的甲基丙烯酸酯类共聚物及医用光学高聚物。
背景技术:
2.自1956年szwarc等报道了阴离子聚合方法后,活性聚合就成为高分子合成方法中最为有效的手段,因其实现了高分子的精确设计以及特定结构和性能的调控。“活性”可控自由基聚合(living radical polymerization/controlled radical polymerization)以其反应条件相对温和、适用单体更广、操作简单、工业成本低等特点和优势,引起了众多科研工作者极大的研究热情和兴趣。“活性”可控自由基聚合包括原子转移自由基聚合(atrp),有机金属自由基聚合(omrp),氮氧化物介导的聚合(nmp)和可逆加成
‑
断裂链转移聚合(raft)等,都为具有明确结构的聚合物的精准构筑提供了可能。
3.由于外界刺激的引入可以实现对聚合行为的调控,如光催化聚合、电催化聚合、机械力控制聚合、氧化还原剂控制聚合等。因此,光作为一种能源被广泛应用于聚合中,与传统的热聚合相比,光聚合可在常温下进行、操作简单、反应平稳,以及可以通过“开”/“关”快速的启动和停止反应等优点。此外,光聚合反应的活化能低,适用于温敏性单体的聚合。
4.在传统的自由基聚合中,大多是通过过渡金属催化剂[即cu(i),ru(ii),fe(ii)]介导休眠种与活性传播的自由基之间的平衡,以维持较低的活性自由基物种浓度。要最小化双分子终止并建立受控/活性链增长显得至关重要。然而,过渡金属催化剂的使用将在最终聚合产物中造成金属污染和残留,会加速聚合物的老化,这也严重限制其在生物医药和电子半导体等行业的应用。
[0005]
因此,如何提供一种无金属污染和残留的共聚反应方案,将其产物用于医用光学高聚物中是具有积极现实意义的课题。
技术实现要素:
[0006]
有鉴于此,本发明的目的在于提出一种实施可靠、合成操作容易且无金属污染残留的基于光催化的甲基丙烯酸酯类共聚物及医用光学高聚物。
[0007]
为了实现上述的技术目的,本发明所采用的技术方案为:
[0008]
二氧杂蒽嵌蒽类化合物作为光敏剂或光催化剂在甲基丙烯酸酯类单体活性自由基聚合中的应用,其中,所述二氧杂蒽嵌蒽类化合物的结构式为式一或式二所示:
[0009][0010]
作为一种可能的实施方式,进一步,所述甲基丙烯酸酯类单体包括:甲基丙烯酸甲酯、甲基丙烯酸丙酯、甲基丙烯酸丁酯、甲基丙烯酸羟乙酯、甲基丙烯酸羟丁酯中的一种以上。
[0011]
本发明还提供一种基于光催化的甲基丙烯酸酯类共聚物,其由甲基丙烯酸酯类单体、二氧杂蒽嵌蒽类催化剂和引发剂混合后,在预设波长的光源辐照下催化共聚合成制得。
[0012]
作为一种可能的实施方式,进一步,所述甲基丙烯酸酯类单体和引发剂的摩尔比例为1~10000∶1,所述二氧杂蒽嵌蒽类催化剂的用量为0.001ppm~1000ppm。
[0013]
作为一种可能的实施方式,进一步,所述反应的温度为
‑
100~100℃,一般情况下,可在室温下直接进行反应合成,本方案的基于光催化的甲基丙烯酸酯类共聚物在反应过程中体系温和。
[0014]
作为一种可能的实施方式,进一步,所述光源的波长为300nm~1000nm。
[0015]
作为一种可能的实施方式,进一步,所述引发剂为包含至少一个c
‑
x键、n
‑
x键、s
‑
x键、o
‑
x键、硫氰酸酯基基
‑
scn、黄原酸酯基
‑
s(c=s)or、三硫代碳酸酯基
‑
s(c=s)sr或硫代氨基甲酸酯基
‑
s(c=o)nrr’;
[0016]
还包括α
‑
溴代苯基乙酸烷基酯、2
‑
溴丙酸烷基酯、2
‑
溴异丁酸烷基酯、溴代丙二酸二烷基酯、2
‑
溴
‑2‑
甲基丙二酸二烷基酯或4
‑
氰基
‑4‑
[(烷基磺酰硫代羰基)磺酰]
‑
戊酸;
[0017]
其中,x为卤素元素f、cl、br或i;
[0018]
另外,所述的烷基、r、r’为c1~c24烷基,包括甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、己基或环己基。
[0019]
作为一种可能的实施方式,进一步,所述二氧杂蒽嵌蒽类催化剂的结构式为:
[0020]
设为pxx1
[0021]
或
[0022]
设为pxx2
[0023]
或
[0024]
设为pxx3
[0025]
或
[0026]
设为pxx4。
[0027]
基于上述方案,本发明还提供基于光催化的甲基丙烯酸酯类共聚物合成方法,其通过将甲基丙烯酸酯类单体、二氧杂蒽嵌蒽类催化剂(即pxx类催化剂)、引发剂和溶剂混合,再对其进行脱气除氧后,将其导入至充有惰性气体的透明反应器中,然后管尾进行液封处理,在预设温度下,通过预设波长的光源对透明流式反应器进行辐照,令其在光源辐照下催化发生共聚合成反应,反应制得的产物经分离后,再通过减压干燥至恒重,获得甲基丙烯
酸酯类共聚物。
[0028]
其中,流动反应器的光源可以为紫外光、可见光或近红外光,只要包含300nm至1000nm区间任意波长的光源即可;装置的供料端可以采用注射器泵供料;反应装置的管路选取以聚四氟乙烯为材料的透明软管。
[0029]
本方案甲基丙烯酸酯类共聚物合成方法的实施方法需要提前排除反应物和反应管中的氧气。将单体,引发剂,光催化剂,溶剂放入带有搅拌的注射器泵中,根据需要可以预先通过冷冻解冻泵循环脱气除氧,向聚四氟乙烯管中充入惰性气氛保护后,将反应液注射进聚四氟乙烯管,管尾接乙醚液封。在一定温度下,选择合适的人工光源或者直接采用太阳光照射来引发聚合。聚合物自身可以沉降或者将反应液倾入不良溶剂(常用乙醚)中沉降分离,后经过洗涤干燥,得到聚合物产品。聚合物分析,可以取干燥的聚合物配成四氢呋喃溶液(浓度1
‑
1.5mg/ml)并通过注射过滤器,过滤后的溶液由gpc测得聚合物的分子量及多分散性。实施实例中所使用的紫色灯泡光源的最大发射在400nm,波长范围为375nm
‑
425nm,蓝色灯泡光源的最大发射在460nm,波长范围为435
‑
485nm。
[0030]
本方案还包括链延长实验,即聚合反应前,将已经提前制备好的大分子引发剂(例如聚甲基丙烯酸甲酯(pmma
‑
br))充分干燥好,避光保存,准确称量好,按照优化的比例条件以此加入单体,pxx类光催化剂和溶剂,混合再通过冷冻解冻泵循环将反应混合物脱气,置于注射器泵,密封,通氩气,使得聚合在惰性气氛中进行,注射进行反应。
[0031]
基于上述的共聚物方案,本发明还提供一种医用光学高聚物,其包括上述所述的基于光催化的甲基丙烯酸酯类共聚物。
[0032]
采用上述的技术方案,本发明与现有技术相比,其具有的有益效果为:本发明方案采用二氧杂蒽嵌蒽类(pxx)化合物作为有机光催化剂在自由基共聚合中的高效催化能力,可以实现甲基丙烯酸甲酯与甲基丙烯酸羟乙酯在不同比例下在可见光调控下的活性自由基聚合,具有催化效率高,易操作易重复等优点;另外,使用取代pxx衍生物作为光催化剂或光敏剂,其在激发后具有较强的还原性,而有机催化剂在光源照射下变成激发态,处于激发态的催化剂用于还原溴化物或硫化物产生自由基和溴或含硫负离子,自由基用于引发单体聚合形成链的增长,随后阴离子和活性链自由基(活性增长链)反应,再转移出电子使得有机光催化剂回到基态,同时活性自由基链在去活化后形成大分子休眠种;如此循环,通过激化活化和去活化,保持休眠种和活性增长链之间的可逆平衡使得聚合反应体系具有一定的可控性,得到分子量可控,分子量分布窄的均聚物或嵌段共聚物。本发明提供的制备方法简单易行,易操作和重复,具有较强的实用性。同时,本方案基于光催化的甲基丙烯酸酯类共聚物具有高纯度和无金属残留的特性,可以用于医用光学材料。
附图说明
[0033]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0034]
图1是本方案流式反应器的现场安装级工作时的影像图;
[0035]
图2是本方案流式反应器的简要反应液流动示意简图;
[0036]
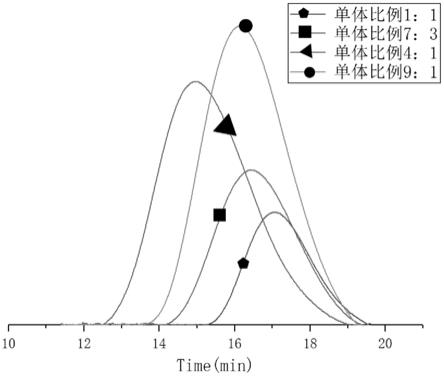
图3是本方案实施例中不同单体比例下的gpc曲线图;
[0037]
图4是本方案实施例中低催化剂用量下大分子引发剂制备的一级动力学图;
[0038]
图5是本方案实施例中在mma:hema为8:2时不同溶剂条件下的gpc曲线图;
[0039]
图6是本方案实施例子中单体比例为1:1得到的甲基丙烯酸甲酯与甲基丙烯酸羟乙酯共聚物的核磁氢谱图;
[0040]
图7是本方案实施例中单体比例为7:3得到的甲基丙烯酸甲酯与甲基丙烯酸羟乙酯共聚物的核磁氢谱图;
[0041]
图8是本方案实施实例中制备大分子引发剂采用的光反应器。
具体实施方式
[0042]
下面结合附图和实施例,对本发明作进一步的详细描述。特别指出的是,以下实施例仅用于说明本发明,但不对本发明的范围进行限定。同样的,以下实施例仅为本发明的部分实施例而非全部实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0043]
实施例1
[0044]
将单体mma(甲基丙烯酸甲酯)和hema(甲基丙烯酸羟乙酯)加入至二氯甲烷(dcm)溶剂中,且令二者形成的混合物[m]浓度分数为12m。
[0045]
按照单体、引发剂和催化剂摩尔比[m]:[dbmm]:[pxx1]=100:1:0.5,单体[m]中的组分比例为mma:hema=8:2,分别将上述原料通过冷冻解冻泵循环将反应混合物脱气,继而移至注射器泵中,然后预先在聚四氟乙烯管制成的流式反应器中通入氩气保持氩气氛围,管尾接乙醚液封,再注射反应液(反应液在流式反应器中填充、流动且受光源辐照,最终进入到乙醚溶液中)。
[0046]
在室温下,用磁力搅拌器充分搅拌注射器泵,然后将反应液中的混合物注入到聚四氟乙烯管中,并通过紫色灯泡(6w)照射(控制反应管中心到光源的距离为10cm)混合物(反应装置如图1或图2所示),直至反应结束;
[0047]
将反应结束的溶液加入到快速搅拌的乙醚溶液中进行沉降处理,将所得沉淀物减压干燥至恒重,得到白色粉末,即甲基丙烯酸酯类共聚物。
[0048]
本实施例中,通过更换光源,平行在蓝色灯泡(6w)照射(控制反应管中心到光源的距离为10cm)混合物的条件下进行再做一组反应。
[0049]
表征检测
[0050]
取紫外和蓝光条件下制得的聚合物,将干燥的聚合物(白色粉末)配成四氢呋喃溶液(浓度1
‑
1.5mg/ml)并通过注射器过滤器,过滤后的溶液由gpc测得聚合物的分子量及多分散性。
[0051]
经测试得到,在本实施例配比条件下,聚合物分子量随着转化率的增长而线性增加。
[0052]
在紫光led照射6h,单体转化率达到91.5%,聚合产物的mn=9.0kda,pdi=1.45;
[0053]
在蓝光led照射6h,单体转化率达到95.4%,聚合产物的mn=9.3kda,pdi=1.39。
[0054]
实施例2
[0055]
将单体mma与hema加入至溶剂(n,n
‑
二甲基乙酰胺,也可为n,n
‑
二甲基甲酰胺、四
氢呋喃、二氯甲烷)中,且令二者形成的混合物[m]浓度分数为12m。
[0056]
按照摩尔比[m]:[dbmm]:[pxx1]=100:1:0.5,单体比例为mma:hema=8:2,分别将上述原料通过冷冻解冻泵循环将反应混合物脱气,移至注射器泵中,预先在聚四氟乙烯管中通入氩气保持氩气氛围,再注射反应液,使得聚合在蓝光下,惰性气氛中进行聚合反应,获得聚合产物,即甲基丙烯酸酯类共聚物。其他操作均与实施例1相同,便不再赘述。
[0057]
本对比例分别通过改变溶剂制得多组聚合产物,将本对比例产物进行表征后,获得如下结果:
[0058]
当体系溶剂换成n,n
‑
二甲基乙酰胺(dma),反应12h,具有91.5%的转化,得到mn=7.4kda的聚合物(pdi=1.49)。
[0059]
溶剂为n,n
‑
二甲基甲酰胺(dmf)时,反应12h,具有90.5%的转化,得到mn=8.4kda的聚合物(pdi=1.32)。
[0060]
溶剂为四氢呋喃时,反应12h,具有92.2%的转化,得到mn=11.4kda的聚合物(pdi=1.47)。
[0061]
溶剂为二氯甲烷时,反应12h,具有92.2%的转化,得到mn=9.8kda的聚合物(pdi=1.37)。
[0062]
如上对比例说明二氯甲烷作为溶剂中的控制是明显优于n,n
‑
二甲基乙酰胺等其他溶剂的,溶剂的选择对聚合反应的控制能力显得十分重要。
[0063]
图5是本方案实施例中在mma:hema为8:2时不同溶剂条件下的gpc曲线图。
[0064]
实施例3
[0065]
本实施例与实施例1大致相同,其不同之处在于,本实施例通过催化剂的种类,分别使用pxx1、pxx2、pxx3、pxx4进行参与反应,所获得的产物经表征获得如下结果:
[0066]
当体系催化剂换成pxx1,反应6h,具有91.5%的转化,得到mn=10.1kda的聚合物(pdi=1.34)。
[0067]
催化剂换成pxx2时,反应6h,具有90.5%的转化,得到mn=9.4kda的聚合物(pdi=1.32)。
[0068]
催化剂换成pxx3时,反应6h,具有90.2%的转化,得到mn=9.2kda的聚合物(pdi=1.37)。
[0069]
催化剂换成pxx4时,反应6h,具有90.2%的转化,得到mn=8.4kda的聚合物(pdi=1.37)。
[0070]
经对比,可说明pxx1作为催化剂,得到的高聚物性能是明显优于其他催化剂的,催化剂的选择对聚合物的性能显得十分重要。
[0071]
实施例4
[0072]
本实施例与实施例1大致相同,其不同之处在于,本实施例为原料加入到10mlschlenk管中,密封,再通过冷冻解冻泵循环将反应混合物脱气,使得聚合在惰性气氛中进行。
[0073]
在室温下,用磁力搅拌器充分搅拌反应混合物,并通过紫色led(6w)照射(控制反应管中心到光源的距离为2cm)混合物。
[0074]
本实施例所获得的产物经表征获得如下结果:
[0075]
在本实施例条件下,在紫光led照射6h,单体转化率达到81.5%,聚合产物的mn=
7.0kda,pdi=1.35。
[0076]
相对实施例1来说,本实施例方案与流式反应器相比转化效率差,这是因为流式反应器采用的聚四氟乙烯管更细小,透光会比反应管更好一些。流式反应器设计的专用液封装置也省去了后处理的繁琐步骤,以及能大量反应,对比在反应管里反应,上述特点流式反应器的优势所在。
[0077]
实施例5
[0078]
本实施例与实施例1大致相同,其不同之处在于,单体[m]中的组分比例为mma:hema=9:1,且反应在蓝光下辐照,其余均与实施例1相同,便不再赘述。
[0079]
本实施例所获得的产物经表征获得如下结果:
[0080]
当单体比例(mma:hema)为9:1,反应6h时,具有92.5%的转化,得到mn=8.8kda的聚合物(pdi=1.34)。
[0081]
实施例6
[0082]
本实施例与实施例1大致相同,其不同之处在于,单体[m]中的组分比例为mma:hema=7:3,且反应在蓝光下辐照,其余均与实施例1相同,便不再赘述。
[0083]
本实施例所获得的产物经表征获得如下结果:
[0084]
当单体比例(mma:hema)为7:3,反应6h时,具有91.5%的转化,得到mn=9.9kda的聚合物(pdi=1.31)。
[0085]
图7是实施例中单体比例为7:3得到的甲基丙烯酸甲酯与甲基丙烯酸羟乙酯共聚物的核磁氢谱图。
[0086]
实施例7
[0087]
本实施例与实施例1大致相同,其不同之处在于,单体[m]中的组分比例为mma:hema=1:1,且反应在蓝光下辐照,其余均与实施例1相同,便不再赘述。
[0088]
本实施例所获得的产物经表征获得如下结果:
[0089]
当单体比例(mma:hema)为1:1,反应6h时,具有88.5%的转化,得到mn=9.1kda的聚合物(pdi=1.29)。
[0090]
其中,图3示出了4~7的不同单体比例下的gpc曲线图。
[0091]
图6是实施例子中单体比例为1:1得到的甲基丙烯酸甲酯与甲基丙烯酸羟乙酯共聚物的核磁氢谱图。
[0092]
pmma
‑
br大分子引发剂的制备
[0093]
将mma(0.50ml,4.7mmol,10,0eq.),dbmm(18μl,94μmol,2eq.)和光催化剂pxx1(2.35μmol,0.05eq.)溶解在0.50ml dcm中,将上述原料加入到10mlschlenk管中,密封,再通过冷冻解冻泵循环将反应混合物脱气,使得聚合在蓝光下,惰性气氛中进行。其他操作参照实施例1。
[0094]
反应6h后,除去反应物,倒入400ml乙醚中并搅拌3h。然后通过真空过滤分离所得沉淀物并用过量乙醚洗涤。然后将聚合物再溶于最少量的dcm中,再次倒入200ml乙醚中并搅拌3h。通过真空过滤再次收集产物,并在减压下干燥,得到白色粉末,即获得pmma
‑
br大分子引发剂(聚合物核磁如图4)(mn=8.10kda,pdi=1.19)。
[0095]
图8是本方案实施实例中制备大分子引发剂采用的光反应器。
[0096]
实施例8
[0097]
将1.42g上述制得的pmma(pmma
‑
br)大分子引发剂(mn=8.10kda,1.0eq.)溶解在12ml的dcm中,再加入5.8ml的hema(3.4
×
10
‑
2mol,202eq.),pxx 1(1.69
×
10
‑
7mol,0.001eq.),根据上述一般聚合步骤在蓝光下反应6h,其他操作参照实施例1。
[0098]
反应6h后,滴加到100ml乙醚中并搅拌5h,通过真空过滤分离所得沉淀物并用过量乙醚洗涤。然后将聚合物再溶于最少量的dcm中,再次倒入50ml乙醚中并搅拌2h,并在真空烘箱中干燥直至在30℃下恒重。
[0099]
经检测,所得的pmma
‑
b
‑
hema共聚物的mn=11.2kda,pdi=1.50。
[0100]
实施例9
[0101]
将1.42g上述制得的pmma(pmma
‑
br)大分子引发剂(mn=8.40kda,1.0eq.)溶解在12ml的dcm中,再加入5.8ml的hema(3.4
×
10
‑
2mol,202eq.),pxx 1(1.69
×
10
‑
7mol,0.001eq.),根据上述一般聚合步骤在蓝光下反应6h,其他操作参照实施例1。
[0102]
反应6h后,滴加到100ml乙醚中并搅拌5h,通过真空过滤分离所得沉淀物并用过量乙醚洗涤。然后将聚合物再溶于最少量的dcm中,再次倒入50ml乙醚中并搅拌2h,并在真空烘箱中干燥直至在30℃下恒重。
[0103]
经检测,所得的pmma
‑
b
‑
hema共聚物的mn=12.2kda,pdi=1.51。
[0104]
实施例10
[0105]
将1.42g上述制得的pmma(pmma
‑
br)大分子引发剂(mn=8.40kda,1.0eq.)溶解在12ml的dcm中,再加入5.8ml的hema(3.4
×
10
‑
2mol,202eq.),pxx1(1.69
×
10
‑
7mol,0.001eq.),根据上述一般聚合步骤在蓝光下反应6h,其他操作参照实施例1。
[0106]
反应6h后,滴加到100ml乙醚中并搅拌5h,通过真空过滤分离所得沉淀物并用过量乙醚洗涤。然后将聚合物再溶于最少量的dcm中,再次倒入50ml乙醚中并搅拌2h,并在真空烘箱中干燥直至在30℃下恒重。
[0107]
经检测,所得的pmma
‑
b
‑
hema共聚物的mn=12.2kda,pdi=1.50。
[0108]
实施例12
[0109]
将1.42g上述制得的pmma(pmma
‑
br)大分子引发剂(mn=8.40kda,1.0eq.)溶解在12ml的dcm中,再加入5.8ml的hema(3.4
×
10
‑
2mol,202eq.),pxx1(1.69
×
10
‑
7mol,0.001eq.),根据上述一般聚合步骤在蓝光下反应6h,其他操作参照实施例1。
[0110]
反应6h后,滴加到100ml乙醚中并搅拌5h,通过真空过滤分离所得沉淀物并用过量乙醚洗涤。然后将聚合物再溶于最少量的dcm中,再次倒入50ml乙醚中并搅拌2h,并在真空烘箱中干燥直至在30℃下恒重。
[0111]
经检测,所得的pmma
‑
b
‑
hema共聚物的mn=14.2kda,pdi=1.60。
[0112]
本方案中,所用pxx(1~4)催化剂的核磁数据如下:
[0113]
pxx 1:
[0114]1h nmr(400mhz,cdcl3)δ7.30(d,j=9.2hz,2h),7.09
‑
7.08(m,4h),6.91(d,j=9.2hz,2h),6.64(t,j=4.4hz,2h)。
[0115]
pxx2:
[0116]1h nmr(500mhz,dmso):δ7.69(d,j=5.9hz,4h),7.57
‑
7.56(m,4h),7.45(t,j=6.3hz,4h),7.37
‑
7.34(m,2h),7.15(d,j=7.5hz,2h),7.01
‑
7.09(m,2h)。
[0117]
pxx3:
[0118]1h nmr(500mhz,dmso):δ7.93(d,j=6.7hz,4h),7.80(d,j=6.9hz,4h),7.67
‑
7.66(m,2h),7.61(d,j=7.6hz,2h),7.18(d,j=7.5hz,2h),7.17
‑
7.16(m,2h)。
[0119]
pxx 4:
[0120]1h nmr(600mhz,cdcl3):δ7.62(d,j=7.8hz,4h),7.51(d,j=8.4hz,4h),7.37(s,2h),7.07
‑
7.11(m,4h),6.62(dd,j=6.8,1.5hz,2h),1.41(s,18h)。
[0121]
以上所述仅为本发明的部分实施例,并非因此限制本发明的保护范围,凡是利用本发明说明书及附图内容所作的等效装置或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

