一种氧化刺槐豆胶
‑
卡拉胶微球及其制备方法
技术领域
1.本发明属于高分子材料领域,具体涉及一种氧化刺槐豆胶
‑
卡拉胶微球及其制备方法。
技术背景
2.刺槐豆胶(lbg)是一种天然的中性半乳甘露聚糖,分子量约30万道尔顿,其结构主要以d
‑
甘露聚糖在1,6位上以β
‑
(1,4)糖苷键连接α
‑
d
‑
吡喃半乳糖,其中半乳糖和甘露糖比例为1:4,是甘露糖含量较多的天然植物籽多糖。据报道,它具有生物相容性、生物吸附性、生物可降解性、非致畸性和非致突变性,表现为粘液粘附行为,其降解产物很容易被排出体外,除了用作增稠剂、稳定剂、乳化剂和胶凝剂之外,也可用于药物制剂和生物医学应用上的赋形剂。
3.目前报道,天然刺槐豆胶生物相容性好,可生物降解,具有耐酸性,较瓜尔胶更好,因此可作为一种潜在的缓释胃溶或肠溶靶向药物输送系统;但由于其黏度高,部分水不溶,导致其利用度不高,因此对其进行改性同时可提高药物负载率,作为一种潜在的药物缓释运输载体。
技术实现要素:
4.为了解决上述的技术问题,本发明提供了一种氧化刺槐豆胶
‑
卡拉胶微球的结构和制备方法,本发明的另一个目的在于提供的微球可作为胃溶型载药介质;
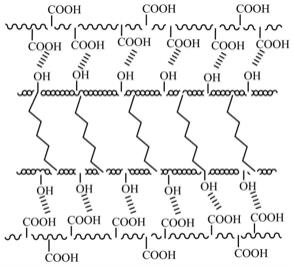
5.所述的氧化刺槐豆胶
‑
卡拉胶微球的结构式如(i)所示:
[0006][0007]
所述的一种氧化刺槐豆胶
‑
卡拉胶微球的制备步骤为:
[0008]
1)取2~10g天然刺槐豆胶在80℃下水合2~6h,转移到反应器中,磁力搅拌下,将反应温度升到45~60℃,加入0.1~1g tempo试剂和0.5~5gnabr搅拌均匀后,将ph为9(2m hcl调节)35ml 10~25%(w/v)的naclo于恒压滴液漏斗中,缓慢滴加,用0.1m等当量的naoh
维持ph为9,滴加完成后继续反应4~7h,加入0.05~0.2g硼氢化钠搅拌30min停止反应,将ph调至8,加入含有nacl的1~2%反应液体积的乙醇终止反应,布氏漏斗过滤,去离子水多次洗涤至中性,用无水乙醇洗涤3次,将固体物质置于50℃下真空干燥,得到固体氧化刺槐豆胶,4~10℃存储;
[0009]
其反应流程如(ii)所示:
[0010][0011]
2)将5g混合比例为1:4的氧化刺槐豆胶和卡拉胶粉末加入至90~150ml蒸馏水中搅拌,磁力搅拌逐渐加热升温至50℃,分散均匀后继续升温至80~90℃,并保温0.5~1h,采用真空脱气装置进行高温脱气,除泡0.5~1h,并降温至50~60℃;将模型药物双氯芬酸钠分散于胶液中形成浓度为25%(w/v)的混合溶液,分散均匀后,加入1~5%(w/v)的戊二醛,反应1~2h得胶粘液,静置24h,得到待用胶液;
[0012]
3)将胶液从21
‑
ga.针头挤入含0.05%~0.1%(w/v)吐温80的90~150ml的氯化钾溶液中(0.1~0.5%(w/v)),将得到的液滴温育10~60min,并通过过滤收集微水凝胶颗粒(i),用两次蒸馏水洗涤过量的表面盐或离子,风干。
[0013]
所述步骤(1)中天然刺槐豆胶与催化氧化体系的添加比为1:0.675~1.12;
[0014]
优选地,天然刺槐豆胶与催化氧化体系的添加比为1:0.995;
[0015]
优选地,所述步骤中所述制备步骤(1)中氧化时间为6h;
[0016]
优选地,所述步骤中所述制备步骤(2)混合溶液中戊二醛浓度为4%(w/v);
[0017]
与现有技术相比,本发明具有如下有益效果:
[0018]
(1)本发明的氧化刺槐豆胶其羧基含量达3200meq/100g,分子量下降,水溶性和生物相容性好;
[0019]
(2)本发明的氧化刺槐豆胶
‑
卡拉胶在戊二醛交联剂下,载药率达68.5%,药物释放时间为28~40min左右。
附图说明
[0020]
图1氧化刺槐豆胶
‑
卡拉胶微球的结构示意图。
具体实施方式
[0021]
为使本发明的目的、技术方案和优点更加清楚明了,下面结合具体实施方式,对本发明进一步详细说明。应该理解,这些描述只是示例性的,而并非要限制本发明的范围。此外,在以下说明中,省略了对公知结构和技术的描述,以避免不必要地混淆本发明的概念。
[0022]
实施例1氧化刺槐豆胶的制备
[0023]
取8g lbg在80℃下水合6h,转移到反应器中,磁力搅拌下,将反应温度升到50℃,加入0.16g tempo试剂和0.8gnabr搅拌均匀后,将ph为9(2m hcl调节)的35ml 10~25%(w/v)的naclo于恒压滴液漏斗中,缓慢滴加,用0.1m等当量的naoh维持ph为9,滴加完成后继续反应6h,加入0.1g硼氢化钠搅拌30min停止反应,将ph调至8,加入含有nacl的2%反应液体积的乙醇终止反应,布氏漏斗过滤,去离子水多次洗涤至中性,用无水乙醇洗涤3次,将固体物质置于50℃下真空干燥,得到氧化刺槐豆胶,4~10℃存储;
[0024]
其中lbg与催化氧化体系的具体添加量见表1;
[0025]
表1:lbg与tempo、nabr、naclo的添加量
[0026][0027]
对上述五组样品进行羧基含量、产物得率、数均分子量(mn)、均重分子量(mw)、多分散指数(pdi),回转半径(rg)进行测定,其测定条件如下:
[0028]
1)羧基含量:按照tappi t237 cm
‑
98(2006)标准测定氧化纤维素中多糖羧基含量。本发明取适量氧化刺槐豆胶,以0.1mol/l盐酸溶液处理2h,随后用去离子水洗涤至滤液呈中性,准确量取定量溶液置于锥形瓶中,迅速向锥形瓶中加入50ml事先配好的nahco3‑
nacl溶液,反应1h后抽滤,取25ml滤液,向其中加入1~2滴甲基红指示剂,随后使用0.01mol/l的盐酸溶液进行滴定。以公式(1)计算羧基含量:
[0029]
(meq/100g)={b
‑
[a (a
×
c/50)]}
×
n
×
(200/w)
ꢀꢀꢀ
(1)
[0030]
其中,a为滴定25ml滤液时盐酸溶液的用量,ml;b为滴定nahco3‑
nacl
[0031]
溶液时盐酸溶液的用量,m l;c为溶液中水的质量,g;n为滴定所用盐酸溶液的实际浓度mol/l;w为样品绝干质量,g;
[0032]
2)氧化刺槐豆胶产物得率:通过过滤收集步骤(1)的水不溶部分,并进行充分洗涤除去杂质,小心转移至密封袋中并称重,取样进行水分测试,氧化产物得率按照公式(2)计算:
[0033]
y(%)=(w2‑
w1)
×
100%
ꢀꢀꢀ
(2)
[0034]
其中,w1为氧化前原料的绝干质量,g;w2为氧化纤维素水不溶部分的绝干质量,g;
[0035]
3)数均分子量(mn)、均重分子量(mw)、多分散指数(pdi),回转半径(rg):采用三重
检测凝胶渗透色谱法(gpc/sec3),在一个模块化系统进行,该系统包括脱气器、hplc泵(k
‑
1001)和ri检测器(k
‑
2300)、粘度计(trisec 270型双检测器)和ralls组成以及两根pl
‑
aquagel
‑
oh凝胶色谱柱(8μm、300
×
7.5mm);纯化过程中洗脱液为0.2mnano3,0.01m nah2po4,0.1%w/v nan3,ph=7,流速为1ml/min;
[0036]
按上述测定方法得到的结果见表2;
[0037]
表2:氧化刺槐豆胶的测定结果
[0038][0039]
lbg与催化氧化体系的添加比如表1结果可知,在氧化过程中,天然刺槐豆胶与催化氧化体系的添加比为1:0.675~1.12,随着催化氧化体系的添加量增加,lbg的氧化程度增加,羧基含量由2.0k增加至3.2k,分子量较空白lbg明显下降,表明氧化过程中,大分子链会解聚,因此分散度和回转半径同时下降,直到lbg与催化氧化体系比为1:0.995(1
‑
4)反应最充分。
[0040]
实施例2氧化刺槐豆胶的制备
[0041]
取8g lbg在80℃下水合6h,转移到反应器中,磁力搅拌下,将反应温度升到50℃,加入0.16g tempo试剂和0.8g nabr搅拌均匀后,将ph为9(2m hcl调节)的35ml 20%(w/v)的naclo于恒压滴液漏斗中,缓慢滴加,用0.1m等当量的naoh维持ph为9,滴加完成后继续反应4、5、6、7h(记为2
‑
1、2
‑
2、2
‑
3、2
‑
4),加入0.1g硼氢化钠搅拌30min停止反应,将ph调至8,加入含有nacl的2%反应液体积的乙醇终止反应,布氏漏斗过滤,去离子水多次洗涤至中性,用无水乙醇洗涤3次,将固体物质置于50℃下真空干燥,得到氧化刺槐豆胶,4~10℃存储;
[0042]
对上述五组样品进行羧基含量、产物得率、数均分子量(mn)、均重分子量(mw)、多分散指数(pdi),回转半径(rg)进行测定,其测定条件同实施例1,得到的结果见表3;
[0043]
表3:氧化刺槐豆胶的测定结果
[0044][0045]
上述五组样品的氧化结果由表2可见,随着氧化时间的增加,羧基含量由1.8k增加至3.2k,分子量较空白lbg明显下降,表明氧化过程中,大分子链会解聚,因此分散度和回转半径同时下降,直到氧化时间为6h反应最充分,继续增加氧化时间,氧化程度不再发生明显变化。
[0046]
实施例3微球的制备
[0047]
将5g混合比例为1:4的氧化刺槐豆胶和卡拉胶粉末加入至100ml蒸馏水中搅拌,磁力搅拌逐渐加热升温至50℃,分散均匀后继续升温至80~90℃,并保温0.5~1h,采用真空脱气装置进行高温脱气,除泡0.5~1h,并降温至50~60℃;将模型药物双氯芬酸钠分散于胶液中形成浓度为25%(w/v)的混合溶液,分散均匀后,分别加入戊二醛形成浓度为的1、2、3、4、5%(w/v)混合溶液(记为3
‑
1、3
‑
2、3
‑
3、3
‑
4、3
‑
5),反应2h得胶粘液,静置24h,将该胶液从21
‑
ga.针头挤入含0.05%(w/v)吐温80的100ml的氯化钾溶液中0.2%(w/v),将液滴温育45min,并通过过滤收集微水凝胶颗粒(i),用两次蒸馏水洗涤过量的表面盐或离子,风干;
[0048]
对上述五组样品进行药物负载率以及完全释放时间考察,其测定条件如下:
[0049]
1)药物负载率:将10mg微球颗粒粉碎,放入100ml的模拟胃液中过夜,使负载药物完全溶解,悬浮液过滤,滤液在276nm处用紫外分光光度计分析,根据式(3)计算药物负载率,取三次平均值:
[0050][0051]
2)完全释放时间:测试微水凝胶在模拟胃液中的即刻释放行为,将50mg样品放入500ml 0.1n的hcl溶液(ph=1.5)中,(50rpm、37
±
0.5℃),记录粒子完全崩解所需的时间,取三次平均值。
[0052]
按上述测定方法得到的测定结果见表4;
[0053]
表4:氧化刺槐豆胶
‑
卡拉胶微球的测定结果
[0054][0055]
戊二醛的添加使得氧化刺槐豆胶与卡拉胶的交联情况可由表3中微球的负载率反映,实施例3中戊二醛的添加量为1~5%,随着戊二醛含量增加,药物负载率由38.5%增加至68.5%,且药物在模拟胃液中的释放时间由28min增加至40min,表明戊二醛使得体系的交联程度增加,药物负载量大且牢固,在酸性模拟胃液中微球崩解的速率下降,可作为胃溶型缓释药物载体。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。