1.本发明涉及一种改性氮化硼、含其的复合材料、其制备方法及应用。
背景技术:
2.电路集成化和微型化使器件内部的热流密度急剧上升,为了实现器件高效稳定的工作,有效的散热系统将成为解决问题的关键。聚合物基导热复合材料以其轻质、高强度、易加工等特点被作为散热器件广泛应用。由于聚合物本身热稳定性较差,其长期处于高热流密度环境下,将会带来巨大的火灾安全隐患,因此在保证高导热的同时,提升复合材料的阻燃性对实际生产和应用具有非常重要的意义。
3.二维层状六方氮化硼由于其特殊的键结构,使其具有优异的热力学性能、机械性能、绝缘性和化学稳定性。理论研究表明单层六方氮化硼面内热导率可达400w/m
·
k,因此,其被做作为导热填料广泛应用在导热复合材料中。在阻燃方面,氮化硼的抗氧化性能非常优异,其二维结构对燃烧过程产生的可燃气体和氧气也具有一定的阻隔能力,但是由于其成碳和抑烟能力较差,因此需要对其进行进一步阻燃改性以提高其综合阻燃性能,同时,也可以提高其界面相容性实现导热增强。
4.由于氮化硼表面缺乏活性基团,因此对其改性的方法多集中在表面官能团的引入(中文专利文献cn103059567a)。首先在空气中煅烧氮化硼,然后在溶剂存在下加入γ―氨丙基三乙氧基硅烷,再进行磷腈修饰得到具有阻燃性的氮化硼。但是在制备过程中,煅烧给予氮化硼表面的活性位点较少,最终导致引入表面的聚磷腈量有限,阻燃性能会受到一定影响。qiu等人利用静电和氢键作用力将植酸类超分子引入氮化硼表面实现阻燃增强(shuilai qiu,yanbei hou,et al.chemical engineering journal 349(2018)223
–
234),由于静电氢键作用力较弱,吸附在氮化硼表面的阻燃剂易脱落,导致综合性能降低。
5.此外,氮化硼本身极性较弱,因此如何在复合材料内部实现其良好的分散性是一个亟待解决的问题。
6.因此,需要开发一种氮化硼表面阻燃改性的方法,阻燃剂在氮化硼表面可稳定且大量存在,在保证环氧树脂优异的综合性能下,赋予环氧树脂复合材料良好的导热和阻燃性能。
技术实现要素:
7.本发明所要解决的技术问题在于,为了克服现有技术中改性的氮化硼表面接枝率较低或者不稳定,其用于复合材料时阻燃性能增强效果不明显的缺陷,因此,本发明提供一种改性氮化硼、含其的复合材料、其制备方法和应用。本发明的改性氮化硼表面充分官能化,能够引入大量阻燃剂,同时可以极大地提升其与环氧树脂的相容性,使复合材料具有良好的导热、机械性能的同时,阻燃性能也大幅度提高,可以满足聚合物基导热复合材料对热稳定性以及阻燃性能的要求。该材料可进一步提高电子器件工作效率以及稳定性,极大地降低器件的火灾风险。
8.为了实现上述目的,本发明提供了一种dopo改性氮化硼的制备方法,其包括:
9.s3.在溶剂存在下,将溴化聚多巴胺改性氮化硼与甲基丙烯酸缩水甘油酯单体、催化体系进行arget atrp(即通过电子转移生成催化剂的原子转移自由基聚合)反应,得到聚甲基丙烯酸缩水酯(pgma)改性的氮化硼,再与9,10
‑
二氢
‑9‑
氧杂
‑
10
‑
磷杂菲
‑
10
‑
氧化物(dopo)混合进行反应,得到dopo改性氮化硼。
10.所述dopo改性氮化硼的制备方法中,所述s3优选包括如下步骤:
11.步骤1:将所述溴化聚多巴胺改性氮化硼在所述溶剂中超声分散,依次与甲基丙烯酸缩水甘油酯单体、溴化亚铜(cubr)、溴化铜(cubr2)、五甲基二乙烯三胺(pmdeta)和抗坏血酸混合,30
‑
40℃(优选30℃)下反应3
‑
4h(优选4h);步骤2:在110
‑
120℃(优选120℃)温度下,将步骤1得到的混合物与9,10
‑
二氢
‑9‑
氧杂
‑
10
‑
磷杂菲
‑
10
‑
氧化物(dopo)进行反应。
12.所述dopo改性氮化硼的制备方法中,所述s3中,所述溶剂为本领域常规极性溶剂,例如所述溶剂选自四氢呋喃、丙酮和n,n
‑
二甲基甲酰胺中的一种或多种,优选n,n
‑
二甲基甲酰胺(dmf),更优选无水dmf。
13.所述dopo改性氮化硼的制备方法中,所述s3中,所述溶剂与所述溴化聚多巴胺改性氮化硼的体积质量比优选40
‑
70ml/g,更优选50ml/g。
14.所述dopo改性氮化硼的制备方法中,所述s3中,所述9,10
‑
二氢
‑9‑
氧杂
‑
10
‑
磷杂菲
‑
10
‑
氧化物(dopo)与所述甲基丙烯酸缩水甘油酯单体的摩尔比优选为(1
‑
2):1,更优选为1:1。
15.所述dopo改性氮化硼的制备方法中,所述s3中,所述溴化聚多巴胺改性氮化硼优选先经超声分散于溶剂中再进行反应。
16.所述dopo改性氮化硼的制备方法中,所述s3中,所述arget atrp反应为本领域常规的arget atrp反应,其反应条件可参考本领域常规arget atrp反应条件进行选择。
17.所述dopo改性氮化硼的制备方法中,所述s3中,所述催化体系为:
18.催化体系1:溴化亚铜、溴化铜、五甲基二乙烯三胺(pmdtea)和抗坏血酸;
19.或,催化体系2:溴化亚铜、溴化铜、1,1,4,7,10,10
‑
六甲基三亚乙基四胺(hmteta)和抗坏血酸。
20.所述dopo改性氮化硼的制备方法中,所述s3中,所述溴化亚铜、溴化铜、五甲基二乙烯三胺和抗坏血酸的摩尔比优选1.9:0.1:(1.9
‑
9.5):(1.9
‑
9.5),更优选1.9:0.1:4:2。
21.所述dopo改性氮化硼的制备方法中,所述s3中,所述溴化亚铜与所述溴化聚多巴胺改性氮化硼的摩尔比优选为1:(5
‑
15),更优选为1:10。
22.所述dopo改性氮化硼的制备方法中,所述s3中,所述arget atrp反应的反应时间优选为3
‑
4小时。
23.所述dopo改性氮化硼的制备方法中,所述s3中,所述arget atrp反应的反应温度优选为30
‑
40℃。
24.所述dopo改性氮化硼的制备方法中,所述s3中,所述聚甲基丙烯酸缩水酯(pgma)改性的氮化硼可通过溴化聚多巴胺改性氮化硼和甲基丙烯酸缩水甘油酯单体的质量比进行调控接枝量从而得到具有不同链长的聚甲基丙烯酸缩水酯(pgma)改性的氮化硼。较佳地,所述溴化聚多巴胺改性氮化硼与所述甲基丙烯酸缩水甘油酯单体的质量比优选为1:(1
‑
50),更优选1:1、1:5、1:10,1:50例如1:1。
25.所述dopo改性氮化硼的制备方法中,所述s3反应结束后,还优选包括后处理步骤:将反应体系离心(例如5000r/min),洗涤沉淀(例如dmf洗涤一次,乙醇洗涤二次,干燥(例如真空烘箱80℃烘6h)后获得dopo改性氮化硼(fbn
‑
dopo)。
26.所述dopo改性氮化硼的制备方法中,所述s3中所述的反应的进程可以通过本领域常规方法进行监控(例如ft
‑
ir、tga、nmr等)。本领域技术人员可依据监测结果(包括原料转化程度、杂质生成情况等)确定何时终止反应,以获得较佳的反应结果。dopo加入后的反应的反应时间可以为8
‑
12小时(例如12小时)。
27.所述dopo改性氮化硼的制备方法中,所述arget atrp反应后,较佳地,不经后处理直接对所述pgma改性的氮化硼进行dopo改性反应。
28.所述dopo改性氮化硼的制备方法中,较佳地,还包括以下步骤:
29.s2.在溶剂、缚酸剂和溴化试剂的存在下,将聚多巴胺改性氮化硼溴化,得到所述溴化聚多巴胺改性氮化硼(fbn
‑
br)。
30.所述dopo改性氮化硼的制备方法中,所述s2优选包括如下步骤:
31.步骤1:将所述聚多巴胺改性氮化硼在所述溶剂中超声分散,与所述缚酸剂混合;
32.步骤2:将所述溴化试剂的溶液与步骤1的反应液混合,在0
‑
4℃(例如0℃)下反应1
‑
4小时(例如1、4小时),再在20
‑
30℃(例如25℃)温度下反应8
‑
12小时(例如8、12小时)。
33.所述dopo改性氮化硼的制备方法中,所述s2中,所述溶剂为本领域常规极性溶剂,例如所述溶剂选自四氢呋喃、丙酮和n,n
‑
二甲基甲酰胺中的一种或多种,优选n,n
‑
二甲基甲酰胺(dmf),更优选无水dmf。
34.所述dopo改性氮化硼的制备方法中,所述s2中,所述溶剂的用量为使所述聚多巴胺改性氮化硼完全分散,优选所述溶剂与所述聚多巴胺改性氮化硼的体积质量比为65
‑
80ml/g,更优选75ml/g。
35.所述dopo改性氮化硼的制备方法中,所述s2中,所述缚酸剂为本领域常规的缚酸剂,优选三乙胺。
36.所述dopo改性氮化硼的制备方法中,所述s2中,所述溴化试剂优选采用可以引发atrp的酰溴,较佳地为2
‑
溴异丁酰溴。
37.所述dopo改性氮化硼的制备方法中,所述s2中,所述溴化试剂与所述聚多巴胺改性氮化硼的质量比优选为1:1
‑
1:2,例如为1:1.5。
38.所述dopo改性氮化硼的制备方法中,所述s2中,所述聚多巴胺改性氮化硼优选经超声分散在溶剂后再进行溴化。
39.所述dopo改性氮化硼的制备方法中,所述s2中,优选步骤1中,所述超声时间一般为将所述聚多巴胺改性氮化硼均匀分散于溶剂中即可,优选5
‑
15min,例如5min、10min、15min,更例如10min。
40.所述dopo改性氮化硼的制备方法中,所述s2中,优选步骤2中,所述滴加的目的是可防止剧烈反应,保证反应充分,同时控制反应的温度优选在0℃。
41.所述dopo改性氮化硼的制备方法中,所述s2的反应时间优选为3
‑
15小时,例如12小时。
42.所述dopo改性氮化硼的制备方法中,所述s2中,所述溴化试剂的溶液的摩尔浓度优选0.2
‑
0.5mol/l,例如为0.3mol/l。
43.所述dopo改性氮化硼的制备方法中,所述s2中,所述溴化结束后,较佳地还包括后处理步骤:将反应体系离心,用乙醇洗涤沉淀(优选洗涤3次),干燥(例如冷冻干燥)即可。所述离心的速率优选5000r/min。
44.所述dopo改性氮化硼的制备方法中,较佳地,还包括以下步骤:
45.s1.在ph为8
‑
9(优选为8.5)的水溶液中,将氮化硼和多巴胺盐酸盐混合,进行反应,得到所述聚多巴胺改性氮化硼(fbn)。
46.所述dopo改性氮化硼的制备方法中,所述s1中优选在室温(例如为20
‑
30℃,再例如为25℃)条件下反应。
47.所述dopo改性氮化硼的制备方法中,所述s1中较佳地包括:在水中超声分散所述氮化硼后,与三羟甲基氨基甲烷混合,搅拌至溶解,再调节ph至8.5,随后与所述盐酸多巴胺混合反应。
48.所述dopo改性氮化硼的制备方法中,所述s1较佳地,所述氮化硼在与多巴胺盐酸盐混合前,所述氮化硼先在水中进行超声分散。较佳地,所述超声分散目的在于将氮化硼均匀分散于水中,其可根据氮化硼的使用量进行选择,优选时间为30
‑
60min(例如为30min、60min)。
49.所述dopo改性氮化硼的制备方法中,所述s1中,用于分散所述氮化硼的水优选去离子水。
50.所述dopo改性氮化硼的制备方法中,所述s1中,用于分散所述氮化硼的水与所述的氮化硼的体积质量比优选为100
‑
150ml/g,更优选为125ml/g。
51.所述dopo改性氮化硼的制备方法中,所述s1中,所述氮化硼优选为二维片层六方氮化硼,较佳地,所述氮化硼的片层长度为0.5μm
‑
10μm,所述氮化硼的片层厚度20nm
‑
500nm。
52.所述dopo改性氮化硼的制备方法中,所述s1中,所述三羟甲基氨基甲烷与所述氮化硼的质量比优选为(0.1
‑
0.6):1,优选0.15:1、0.3:1或0.6:1,更优选0.6。
53.所述dopo改性氮化硼的制备方法中,所述s1中,所述三羟甲基氨基甲烷可为固体形式或水溶液形式。
54.所述dopo改性氮化硼的制备方法中,所述s1中,较佳地,通过hcl水溶液和三羟甲基氨基甲烷调节ph至8.5。所述hcl水溶液的摩尔浓度优选为0.1
‑
0.5mol/l(例如为0.1mol/l、0.2mol/l、0.5mol/l),更优选为0.1mol/l。
55.所述dopo改性氮化硼的制备方法中,所述s1中,所述盐酸多巴胺与所述氮化硼的质量比优选1:(1
‑
4),更优选1:1、1:2或1:4,最优选1:4。
56.所述dopo改性氮化硼的制备方法中,所述s1中所述的反应的进程本领域技术人员可依据监测结果(包括反应颜色、原料转化程度、tga表面黏附量)确定何时终止反应,以获得较佳的反应结果。所述反应时间可以为10
‑
12h小时(例如12小时)。
57.所述dopo改性氮化硼的制备方法中,所述s1反应结束后,还优选包括后处理步骤:将反应体系离心,水洗涤沉淀(优选洗涤3次),干燥(例如冷冻干燥)后获得所述聚多巴胺修饰的氮化硼(fbn)。所述离心的速率优选为5000r/min。
58.本发明还提供了一种由上述dopo改性氮化硼的制备方法制得的dopo改性氮化硼。
59.本发明还提供了一种pgma改性氮化硼的制备方法,其包括:
60.在溶剂存在下,将溴化聚多巴胺改性氮化硼与甲基丙烯酸缩水甘油酯单体、催化体系进行arget atrp(即通过电子转移生成催化剂的原子转移自由基聚合)反应,得到聚甲基丙烯酸缩水酯(pgma)改性的氮化硼。
61.所述pgma改性氮化硼的制备方法的条件同上述dopo改性氮化硼的制备方法所述。
62.本发明还提供了一种由上述pgma改性氮化硼的制备方法制得的pgma改性氮化硼。
63.本发明还提供了一种含上述的dopo改性氮化硼的复合材料的制备方法,所述含dopo改性氮化硼的复合材料的制备方法包括如下步骤:
64.s5.所述dopo改性氮化硼分散于溶剂中,得dopo改性氮化硼分散液,与环氧树脂预聚体进行反应,得到环氧树脂预聚体/氮化硼分散液;
65.s6.将步骤s5得到的环氧树脂预聚体/氮化硼分散液除去溶剂,与固化剂混合,真空脱泡,固化,得到含所述改性氮化硼的复合材料。
66.所述复合材料的制备方法中,所述s5优选包括如下步骤:室温下(20
‑
30℃),通过搅拌以及超声分散使所述步骤s3中得到的dopo改性氮化硼在溶剂中分散均匀,得到dopo改性氮化硼的分散液,与环氧树脂预聚体混合,搅拌后得到环氧树脂预聚体/氮化硼分散液;更优选,将所述环氧树脂预聚体加入到所述分散液中。
67.所述复合材料的制备方法中,所述s6优选包括如下步骤:将所述步骤s5得到的环氧树脂预聚体/氮化硼分散液,除去溶剂,与固化剂搅拌均匀进行真空脱泡,最后升温固化(例如80℃固化4h,120℃固化6h),得到复合材料,更优选,将所述环氧树脂预聚体/氮化硼分散液除去溶剂,加入固化剂搅拌均匀。
68.所述复合材料的制备方法中,所述s5中,所述溶剂为本领域常规极性溶剂,所述溶剂选自四氢呋喃、丙酮和n,n
‑
二甲基甲酰胺中的一种或多种,较佳地为四氢呋喃(thf)。
69.所述复合材料的制备方法中,所述s5中,所述溶剂与所述环氧树脂预聚体的体积质量比优选0.7
‑
1.2ml/g,更优选0.75ml/g。
70.所述复合材料的制备方法中,所述s5中,所述氮化硼的添加量优选为复合材料的10
‑
30wt%,例如为10wt%、20wt%或30wt%。所述氮化硼的添加量是指纯氮化硼的含量,不包括接枝阻燃剂改性,添加量是相对于复合材料的质量占比。
71.所述复合材料的制备方法中,所述s5中,所述环氧树脂为双酚a型环氧树脂,较佳地,为双酚a缩水甘油醚。
72.所述复合材料的制备方法中,所述s5中,所述环氧树脂预聚体可商业购买获得或采用本领域常规方法制得。
73.所述复合材料的制备方法中,所述s6中,所述固化剂较佳地为咪唑类固化剂、胺类固化剂或酸酐类固化剂。
74.所述复合材料的制备方法中,所述s6中,所述固化剂的用量优选为环氧树脂预聚体的2
‑
50wt%,更优选6wt%。
75.所述复合材料的制备方法中,所述s6中,所述固化分为两个阶段,较佳地,第一阶段为60
‑
90℃(优选80℃),第二阶段为90
‑
150℃(优选120℃);第一阶段固化时间为4
‑
6h(例如4h),第二阶段固化时间为6
‑
8h(例如6h)。
76.所述复合材料的制备方法中,所述s6中,所述真空脱泡为本领域常规的真空脱泡方法。
77.本发明还提供了一种由上述含dopo改性氮化硼的复合材料的制备方法制备得到的复合材料。
78.本发明还提供了一种所述复合材料在制备散热器件中的应用。较佳地,所述散热器件为led灯罩或电子器件封装材料。
79.本发明的积极进步效果在于:本发明的复合材料兼具阻燃性和导热性,加入30wt%改性氮化硼后其热导率可达1.249w/m
·
k,添加20%改性氮化硼,复合材料阻燃性能得到极大的提升,极限氧指数可达到26%,燃烧过程最大放热速率降低近47.31%,总热释放量降低了近44.55%。(热导率)烟释放量也得到了有效抑制。改性氮化硼优异的热稳定性和二维阻隔性能以及阻燃剂对基体的成炭作用,三者共同实现了材料的阻燃增强。
80.本发明对氮化硼表面进行充分的修饰,在其表面引入的阻燃剂分子的官能团可以调控。设计的阻燃剂分子可以匹配树脂基体的官能团,增加了氮化硼和环氧树脂之间的相容性,有利于提高复合材料的力学性能。
81.本发明所制备的导热阻燃环氧树脂复合材料的力学性能有较大幅度提高,20wt%的改性氮化硼加入基体中与未改性的氮化硼相比拉伸强度最大可以提升33%,模量可以提升26%,此外,氮化硼和环氧树脂之间良好的界面相容性降低了它们之间的界面热阻,可以进一步提升复合材料的热导率。
82.相比于添加传统有机阻燃剂增加导热复合材料的阻燃性能,本发明所制备的有机阻燃剂修饰无机氮化硼的有机
‑
无机杂化型阻燃剂作为填料加入到环氧树脂基体中,可以更大地发挥其阻燃以及相容的功能。阻燃改性的氮化硼具有独特的二维片层结构,以及优异的热稳定性,这将使其成为一种高效的有机
‑
无机杂化型阻燃剂。与传统的无机阻燃剂相比,阻燃性氮化硼具有较高的热导率,添加到基体中可以实现复合材料导热性能和阻燃性能的增强。
83.本发明提供的改性氮化硼以及基于改性氮化硼的复合材料具有如下优点:
84.1.氮化硼表面接枝的阻燃剂含量较高且稳定,接枝的阻燃剂分子链的种类和长度都可控调节。
85.2.较低的阻燃剂含量下可以实现较大幅度的阻燃性能增强。
86.3.修饰阻燃剂的氮化硼在复合材料中易分散均匀,并且界面相容性良好,可以大幅提升复合材料的力学性能。
87.4.因为氮化硼本身的高导热性和热稳定性,复合材料的在具有良好阻燃性的同时也具有高的热导率,因此,本发明提供的改性氮化硼以及基于改性氮化硼制备的导热阻燃复合材料具有广泛的生产和应用前景。
附图说明
88.图1是未改性的氮化硼的扫描电镜图
89.图2是溴化聚多巴胺改性氮化硼的x射线光电子能谱图
90.图3是环氧树脂/氮化硼复合材料的断面形貌扫描电镜图,(a)是环氧树脂/10wt%氮化硼断面图、(b)是环氧树脂/20wt%氮化硼断面图,(c)是环氧树脂/10wt%改性氮化硼断面图、(d)是环氧树脂/20wt%改性氮化硼断面图。
91.图4是复合材料的拉伸性能图,(a)是不同氮化硼含量的环氧树脂复合材料的拉伸
强度图,(b)是不同氮化硼含量的环氧树脂复合材料的拉伸模量图。
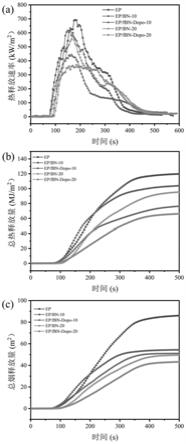
92.图5是复合材料的锥形量热测试,(a)热释放速率与时间的变化关系、(b)总热释放量随时间的变化关系、(c)总烟释放量随时间的变化关系图。
93.图6是复合材料热导率随氮化硼含量变化图。
94.图7是改性氮化硼的热重分析图。
95.图8是改性氮化硼的表面形貌图和电子能谱图。
96.图9是改性氮化硼的傅里叶红外光谱图。
具体实施方式
97.下面通过具体实施例的方式进一步详细说明本发明,但并不因此将本发明限制在所述实施例范围之中。此外,下面描述的本发明各个实施例中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。实施实例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
98.为了制备高导热阻燃复合材料,本发明是以氮化硼为导热填料,然后对其进行阻燃改性。以下各实施例和对比例中,氮化硼的片层长度在0.5
‑
10μm之内,片层厚度在20
‑
500nm之内,如图1所示。氮化硼以及改性氮化硼的填充量在10wt%
‑
30wt%之内,对比例中所用氮化硼为未改性氮化硼,对比例和实施例中所用环氧树脂均为双酚a环氧树脂,固化剂为咪唑类固化剂。
99.本发明中涉及现有的实验试剂的厂家型号具体如下:
100.六方氮化硼platelets cfp 003sf,3m公司;
101.双酚a型环氧树脂(≥85.0%),九鼎化学;
[0102]2‑
乙基
‑4‑
甲基咪唑(≥96.0%),阿拉丁;
[0103]2‑
溴异丁酰溴(≥98%),阿拉丁;
[0104]
甲基丙烯酸缩水甘油酯(≥97%),阿拉丁;
[0105]
盐酸多巴胺(≥98.0%),阿拉丁;
[0106]
9,10
‑
二氢
‑9‑
氧杂
‑
10
‑
磷杂菲
‑
10
‑
氧化物(≥97%),阿拉丁。
[0107]
本发明实施例中,室温即为25℃。
[0108]
实施例1
[0109]
一、改性氮化硼的制备
[0110]
(1)将4g氮化硼加入到500ml去离子水中,室温下(25℃)充分搅拌均匀后超声分散30min;然后加入2.4g三羟甲基氨基甲烷,搅拌充分溶解;随后再加入0.1mol/l的hcl溶液调节ph为8.5;之后,加入1g盐酸多巴胺25℃下搅拌12h;将所得溶液离心(5000r/min)取沉淀,用去离子水洗涤三次,干燥后得到黑色粉末状聚多巴胺修饰的氮化硼,为了方便表达将其简称为fbn。
[0111]
(2)将2g fbn加入到150ml无水n,n
‑
2甲基甲酰胺(dmf)中,充分搅拌均匀后超声分散;随后加入6mmol三乙胺(tea)充分溶解;将6mmol的2
‑
溴异丁酰溴溶解在20ml无水dmf中,然后将其缓慢滴入到fbn分散液中,滴入过程持续10min;随后在冰水浴(即0℃)中搅拌反应4h,室温反应8h;将所得溶液离心(5000r/min)取沉淀,用乙醇洗涤三次,干燥后得到溴化的氮化硼,为了方便表达将其简称为fbn
‑
br。
[0112]
(3)将100mg fbn
‑
br加入到5ml无水dmf中,充分搅拌均匀后超声分散;加入0.1g(0.7mmol)甲基丙烯酸缩水甘油醚,按照1.9:0.1:3:2的比例,加入0.019mmol溴化亚铜(cubr),0.001mmol溴化铜(cubr2),0.03mmol五甲基二乙烯三胺(pmdeta),0.02mmol抗坏血酸,在30℃下反应4h;紧接着,将温度升至120℃,加入0.8mmol的9,10
‑
二氢
‑9‑
氧杂
‑
10
‑
磷杂菲
‑
10
‑
氧化物(dopo),反应12h;结束反应后将所得溶液离心(5000r/min)取沉淀,用dmf洗涤一次,乙醇洗涤二次,干燥后得到阻燃改性的氮化硼,为了方便表达将其简称为fbn
‑
dopo。
[0113]
二、基于阻燃改性氮化硼的复合材料制备
[0114]
(4)取10g fbn
‑
dopo(其中氮化硼的含量约为5.3g)加入30ml四氢呋喃,经过搅拌均匀分散;然后加入40g环氧树脂预聚体(即双酚a缩水甘油醚),机械搅拌混合均匀,得到均匀的环氧树脂预聚物/阻燃氮化硼分散液。
[0115]
(5)将得到的环氧树脂预聚体/阻燃氮化硼分散液,先经过旋转蒸馏除去四氢呋喃,再加入2.4g的2
‑
乙烯
‑4‑
甲基咪唑固化剂,使用行星搅拌器在室温下搅拌10min保证分散均匀;紧接着在50℃下真空脱泡10min,最后将得到的混合液倒入特氟龙模具中,在80℃固化4h,120℃固化6h,得到基于改性氮化硼的环氧树脂复合材料,为了方便表达将其简称为ep/fbn
‑
dopo
‑
10。
[0116]
图1所示为氮化硼的扫描电镜图片,氮化硼的尺寸为0.5
‑
10μm。使用x射线光电子能谱对fbn
‑
br进行表征,其表面元素含量如图2所示,表明其表面可以成功接枝含溴引发位点。
[0117]
如图使用热重分析tga(netzsch sta
‑
449f3)对改性氮化硼进行分析,如图7所示,pda的接枝量相当于氮化硼的质量为13.5%,fbn
‑
dopo中pgma的接枝量相当于氮化硼的质量为18%,dopo的接枝量相当于氮化硼的质量为15%。
[0118]
利用扫描电子显微镜观察了改性以后的氮化硼(fbn
‑
dopo)的表面形貌,如图8所示,改性氮化硼表面出现褶皱层,聚合物被有效包裹上去,通过x射能谱分析(eds)可以证明改性过程是成功的。通过傅里叶红外光谱对fbn
‑
dopo进行表征,如图9所示,与氮化硼相比,fbn
‑
dopo的红外光谱图发生了如下变化,在1725处出现c=o cm
‑1峰,在1152cm
‑1处出现环氧基团特征峰,在1205cm
‑1和754cm
‑1处出现p=o和p
‑
o
‑
ph特征峰,以上证明了pgma和dopo的成功接枝。
[0119]
为了验证fbn
‑
dopo在环氧树脂中的分散性与界面性能,将所制备的ep/fbn
‑
dopo
‑
10复合材料在液氮中脆断,利用扫描电子显微镜观察纳米改性氮化硼在基体中的分布情况以及其与基体的粘结情况。所得结果如图3(c)所示,fbn
‑
dopo在基体中整体分布非常均匀,几乎没有团聚现象,而且,fbn
‑
dopo和基体结合紧密没有出现裂隙。由于fbn
‑
dopo和基体具有较好的界面相容性和较少的界面缺陷,所以在断裂过程中力学性能可以大幅度提升。按照gb/t 1040
‑
2006对ep/fbn
‑
dopo
‑
10的力学性能进行测试,结果如图4所示,得到拉伸强度为32.74mpa,拉伸模量为5912mpa,拉伸强度较纯环氧树脂提升了9.133%,拉伸模量较纯环氧树脂提升了68.99%。这表明表面改性有利于提升氮化硼和环氧树脂的界面相容性,有利于提升复合材料的力学性能。
[0120]
如图6所示,按照astm
‑
c1113 90标准测得ep/fbn
‑
dopo
‑
10的热导率为0.3492w/m
·
k,较纯的环氧树脂(0.201w/m
·
k)提高了73.73%。按照gb/t 2495
‑
2009和gb/t 2408
‑
2008对ep/bn
‑
10测试了极限氧指数和垂直燃烧等级进行测试,极限氧指数从21.5%提高到了23.6%,垂直燃烧等级没有提升。
[0121]
按照iso 5660
‑
1:2002对ep/bn
‑
10进行锥形量热测试,结果表明,ep/fbn
‑
dopo
‑
10复合材料在整个燃烧过程的热释放速率(hrr)均小于环氧树脂,如图5所示,其中峰值热释放速率(phrr)从692.3kw/m2降低到443.7kw/m2,总热释放量(thr)从120.2kw/m2降低到76.71kw/m2,总的发烟量(tsp)从86.40m2降低到51.02m2,上述结果表明复合材料ep/fbn
‑
dopo
‑
10的阻燃性能较环氧树脂有较大的提升。
[0122]
实施例2
[0123]
一、改性氮化硼的制备
[0124]
(1)将4g氮化硼加入到500ml去离子水中,室温下(25℃)充分搅拌均匀后超声分散30min;然后加入2.4g三羟甲基氨基甲烷,搅拌充分溶解;随后再加入0.1mol/l的hcl溶液调节ph为8.5;之后,加入1g盐酸多巴胺25℃下搅拌12h;将所得溶液离心(5000r/min)取沉淀,用去离子水洗涤三次,干燥后得到黑色粉末状聚多巴胺修饰的氮化硼,为了方便表达将其简称为fbn。
[0125]
(2)将2g fbn加入到150ml无水n,n
‑
2甲基甲酰胺(dmf)中,充分搅拌均匀后超声分散;随后加入6mmol三乙胺(tea)充分溶解;将6mmol的2
‑
溴异丁酰溴溶解在20ml无水dmf中,然后将其缓慢滴入到fbn分散液中,滴入过程持续10min;随后在冰水浴(即0℃)中搅拌反应4h,室温反应8h;将所得溶液离心(5000r/min)取沉淀,用乙醇洗涤三次,干燥后得到溴化的氮化硼,为了方便表达将其简称为fbn
‑
br。
[0126]
(3)将100mgfbn
‑
br加入到5ml无水dmf中,充分搅拌均匀后超声分散;加入0.1g(0.7mmol)甲基丙烯酸缩水甘油醚,按照1.9:0.1:3:2的比例,加入0.019mmol溴化亚铜(cubr),0.001mmol溴化铜(cubr2),0.03mmol五甲基二乙烯三胺(pmdeta),0.02mmol抗坏血酸,在30℃下反应4h;紧接着,将温度升至120℃,加入0.8mmol的9,10
‑
二氢
‑9‑
氧杂
‑
10
‑
磷杂菲
‑
10
‑
氧化物(dopo),反应12h;结束反应后将所得溶液离心(5000r/min)取沉淀,用dmf洗涤一次,乙醇洗涤二次,干燥后得到阻燃改性的氮化硼,为了方便表达将其简称为fbn
‑
dopo。
[0127]
二、基于阻燃改性氮化硼的复合材料制备
[0128]
(4)取22.5g fbn
‑
dopo(其中氮化硼的含量约为11.9g)加入30ml四氢呋喃,经过搅拌均匀分散;然后加入35g环氧树脂预聚体(即双酚a缩水甘油醚),机械搅拌混合均匀,得到均匀的环氧树脂预聚物/阻燃氮化硼分散液。
[0129]
(5)将得到的环氧树脂预聚体/阻燃氮化硼分散液,先经过旋转蒸馏除去四氢呋喃,再加入2.1g的2
‑
乙烯
‑4‑
甲基咪唑固化剂,使用行星搅拌器在室温下搅拌10min保证均匀分散;紧接着在50℃下真空脱泡10min,最后将得到的混合液倒入特氟龙模具中,在80℃固化4h,120℃固化6h,得到基于改性氮化硼的环氧树脂复合材料,为了方便表达将其简称为ep/fbn
‑
dopo
‑
20。
[0130]
为了验证fbn
‑
dopo在环氧树脂中的分散性与界面性能,将所制备的ep/fbn
‑
dopo
‑
20复合材料在液氮中脆断,利用扫描电子显微镜观察纳米改性氮化硼在基体中的分布情况以及其与基体的粘结情况。所得结果如图3(d)所示,fbn
‑
dopo在基体中整体分布非常均匀,几乎没有团聚现象,而且,fbn
‑
dopo和基体结合紧密没有出现裂隙。由于fbn
‑
dopo和基体具
有较好的界面相容性和较少的界面缺陷,所以在断裂过程中力学性能可以大幅度提升。
[0131]
按照gb/t 1040
‑
2006对ep/fbn
‑
dopo
‑
20的力学性能进行测试,结果如图4所示,得到拉伸强度为32.61mpa,拉伸模量为8401mpa,拉伸强度较纯环氧树脂提升了8.7%,拉伸模量较纯环氧树脂提升了138.9%。这表明表面改性有利于提升氮化硼和环氧树脂的界面相容性,有利于提升复合材料的力学性能。
[0132]
如图6所示,按照astm
‑
c1113 90标准测得ep/fbn
‑
dopo
‑
20的热导率为0.7509w/m
·
k,较纯的环氧树脂(0.201w/m
·
k)提高了273.6%。按照gb/t 2495
‑
2009和gb/t 2408
‑
2008对ep/fbn
‑
dopo
‑
20测试了极限氧指数和垂直燃烧等级进行测试,极限氧指数从21.5%提高到了26%,垂直燃烧等级提升到v2。
[0133]
按照iso 5660
‑
1:2002对ep/fbn
‑
dopo
‑
20进行锥形量热测试,结果表明,ep/fbn
‑
dopo
‑
20复合材料在整个燃烧过程的热释放速率(hrr)均小于环氧树脂,如图5所示,其中峰值热释放速率(phrr)从692.3kw/m2降低到364.8kw/m2,总热释放量(thr)从120.2kw/m2降低到66.65kw/m2,总的发烟量(tsp)从86.40m2降低到43.28m2,上述结果表明复合材料ep/fbn
‑
dopo
‑
20的阻燃性能较环氧树脂有非常大的提升。
[0134]
实施例3
[0135]
取37.8g实施例1制备得到的fbn
‑
dopo(其中氮化硼的含量约为20g)加入30ml四氢呋喃,经过搅拌均匀分散;然后加入27g环氧树脂预聚体(即双酚a缩水甘油醚),机械搅拌混合均匀,得到均匀的环氧树脂预聚物/阻燃氮化硼分散液。
[0136]
将得到的环氧树脂预聚体/阻燃氮化硼分散液,先经过旋转蒸馏除去四氢呋喃,再加入1.6g的2
‑
乙烯
‑4‑
甲基咪唑固化剂,使用行星搅拌在室温下搅拌10min保证均匀分散;紧接着在50℃下真空脱泡10min,最后将得到的混合液倒入特氟龙模具中,在80℃固化4h,120℃固化8h,得到基于改性氮化硼的环氧树脂复合材料,为了方便表达将其简称为ep/fbn
‑
dopo
‑
30。
[0137]
按照gb/t 1040
‑
2006对ep/fbn
‑
dopo
‑
30的力学性能进行测试,结果如图4所示,得到拉伸强度为29mpa,拉伸模量为11143mpa,拉伸强度较纯环氧树脂降低了3.3%,拉伸模量较纯环氧树脂提升了216.8%。这表明表面改性有利于提升氮化硼和环氧树脂的界面相容性,有利于提升复合材料的力学性能。
[0138]
如图6所示,按照astm
‑
c1113 90标准测得ep/fbn
‑
dopo
‑
30的热导率为1.249w/m
·
k,较纯的环氧树脂(0.201w/m
·
k)提高了521.4%。良好的界面相容性极大的降低了氮化硼和环氧树脂之间的界面热阻,使其热导率有非常大的提升。
[0139]
实施例4:按实施例1步骤(3)中的方法,差别仅在于改变甲基丙烯酸缩水甘油醚的量不同,通过热重测得不同投料比下的接枝量变化。结果见表1:
[0140]
表1不同投料比对应的接枝量
[0141]
投料比热重分析失重量(%)接枝率(%)1:130.918.71:536.424.21:1051.138.91:5085.273
[0142]
表1中的投料比是指:溴化聚多巴胺改性氮化硼与甲基丙烯酸缩水甘油酯单体的
质量比。
[0143]
对比例1
[0144]
制备含有10wt%氮化硼的阻燃导热复合材料,称取5.3g氮化硼在100℃的高温烘箱中烘干6h;称取45g环氧树脂预聚体(即双酚a缩水甘油醚)与烘干的氮化硼混合,使用机械搅拌对上述混合物室温下搅拌5min制得均匀分散的环氧树脂预聚体/氮化硼分散液;将2.7g的2
‑
乙烯
‑4‑
甲基咪唑固化剂滴入上述环氧树脂预聚体/氮化硼分散液中,使用行星搅拌器在室温下再搅拌10min保证分散均匀;紧接着在50℃下真空脱泡10min,最后将得到的混合液倒入特氟龙模具(拉伸、导热、锥形量热测试的标准样条),紧接着在80℃固化4h,120℃固化6h,最终得到环氧树脂/氮化硼复合材料,为了方便表达将其简称为ep/bn
‑
10。
[0145]
为了验证氮化硼在环氧树脂中的分散性与界面性能,将所制备的环氧树脂/氮化硼复合材料在液氮中脆断,断面喷金后利用扫描电子显微镜观察纳米氮化硼在基体中的分布情况以及其与基体的粘结情况。所得结果如图3(a)所示,氮化硼在基体中整体分布均匀,局部区域氮化硼发生团聚,氮化硼纳米片与基体之间的缝隙较宽,表明在断裂过程中与基体发生脱黏,所以未经改性的氮化硼与基体的界面强度较差。
[0146]
按照gb/t 1040
‑
2006对ep/bn
‑
10的力学性能进行测试,结果如图4所示,得到拉伸强度为25.9mpa,拉伸模量为4823mpa,拉伸强度较纯环氧树脂降低了13.67%,拉伸模量较纯环氧树脂提升了37.13%。氮化硼与环氧树脂界面作用力较差,界面存在缺陷,所以拉伸强度出现较为明显的下降。
[0147]
如图6所示,按照astm
‑
c1113 90标准测得ep/bn
‑
10的热导率为0.316w/m
·
k,较纯的环氧树脂(0.201w/m
·
k)提高了57.21%。按照gb/t 2495
‑
2009和gb/t 2408
‑
2008对ep/bn
‑
10测试了极限氧指数和垂直燃烧等级进行测试,极限氧指数从21.5%提高到了22.3%,垂直燃烧等级没有提升。
[0148]
按照iso 5660
‑
1:2002对ep/bn
‑
10进行锥形量热测试,结果表明,ep/bn
‑
10复合材料在整个燃烧过程的热释放速率(hrr)均小于环氧树脂,如图5所示,其中峰值热释放速率(phrr)从692.3kw/m2降低到632.9kw/m2,总热释放量(thr)从120.2kw/m2降低到105.5kw/m2,总的发烟量(tsp)从86.40m2降低到54.28m2,上述结果表明复合材料ep/bn
‑
10的阻燃性能较环氧树脂有一定的提升。
[0149]
对比例2
[0150]
制备含有20wt%氮化硼的阻燃导热复合材料,称取11.9g氮化硼在100℃的高温烘箱中烘干6h;称取45g环氧树脂预聚体(即双酚a缩水甘油醚)与烘干的氮化硼混合,使用机械搅拌对上述混合物室温搅拌50min,制得均匀分散的环氧树脂预聚体/氮化硼分散液;将2.7g的2
‑
乙烯
‑4‑
甲基咪唑固化剂滴入上述环氧树脂预聚体/氮化硼分散液中,使用行星搅拌器在室温下搅拌10min保证均匀分散;紧接着使用高速脱泡机对混合液脱泡处理5min(2000r/min),最后将得到的混合液倒入特氟龙模具(拉伸、导热、锥形量热测试的标准样条),紧接着在80℃固化4h,120℃固化6h,最终得到环氧树脂/氮化硼复合材料,为了方便表达将其简称为ep/bn
‑
20。
[0151]
为了验证氮化硼在环氧树脂中的分散性与界面性能,将所制备的环氧树脂/氮化硼复合材料在液氮中脆断,断面喷金后利用扫描电子显微镜观察纳米氮化硼在基体中的分布情况以及其与基体的粘结情况。所得结果如图3(b)所示,氮化硼在基体中整体分布均匀,
局部区域氮化硼发生团聚,氮化硼纳米片与基体之间的缝隙较宽,表明在断裂过程中与基体发生脱黏,所以未经改性的氮化硼与基体的界面强度较差。
[0152]
按照gb/t 1040
‑
2006对ep/bn
‑
10的力学性能进行测试,结果如图4所示,得到拉伸强度为24.52mpa,拉伸模量为6683mpa,拉伸强度较纯环氧树脂降低了18.27%,拉伸模量较纯环氧树脂提升了90.02%。氮化硼与环氧树脂界面作用力较差,界面存在缺陷,所以拉伸强度出现较为明显的下降。
[0153]
如图6所示,按照astm
‑
c1113 90标准测得ep/bn
‑
20的热导率为0.6521w/m
·
k,较纯的环氧树脂(0.201w/m
·
k)提高了224.4%。按照gb/t 2495
‑
2009和gb/t 2408
‑
2008对ep/bn
‑
20测试了极限氧指数和垂直燃烧等级进行测试,极限氧指数从21.5%提高到了22.8%,垂直燃烧等级没有提升。
[0154]
按照iso 5660
‑
1:2002对ep/bn
‑
20进行锥形量热测试,结果表明,ep/bn
‑
20复合材料在整个燃烧过程的热释放速率(hrr)均小于环氧树脂,如图5所示,与纯环氧树脂相比,峰值热释放速率(phrr)从692.3kw/m2降低到583.8kw/m2,总热释放量(thr)从120.2kw/m2降低到95.96kw/m2,总的发烟量(tsp)从86.40m2降低到57.18m2,上述结果表明复合材料ep/bn
‑
20的阻燃性能较环氧树脂有一定的提升。
[0155]
对比例3
[0156]
制备含有30wt%氮化硼的阻燃导热复合材料,称取20g氮化硼在100℃的高温烘箱中烘干6h;称取45g环氧树脂预聚体(即双酚a缩水甘油醚)与烘干的氮化硼混合,使用机械搅拌对上述混合在室温下搅拌50min,制得均匀分散的环氧树脂预聚体/氮化硼分散液;将2.7g的2
‑
乙烯
‑4‑
甲基咪唑固化剂滴入上述环氧树脂预聚体/氮化硼分散液中,使用行星搅拌器在室温下搅拌10min保证均匀分散;紧接着使用高速脱泡机对混合液脱泡处理5min(2000r/min),最后将得到的混合液倒入特氟龙模具(拉伸、导热、锥形量热测试的标准样条),紧接着在80℃固化4h,120℃固化6h,最终得到环氧树脂/氮化硼复合材料,为了方便表达将其简称为ep/bn
‑
30。
[0157]
按照gb/t 1040
‑
2006对ep/bn
‑
30的力学性能进行测试,结果如图4所示,得到拉伸强度为22.25mpa,拉伸模量为9291mpa,拉伸强度较纯环氧树脂降低了25.83%,拉伸模量较纯环氧树脂提升了164.2%。氮化硼与环氧树脂界面作用力较差,界面存在缺陷,所以拉伸强度出现较为明显的下降。
[0158]
如图6所示,按照astm
‑
c1113 90标准测得ep/bn
‑
30的热导率为1.063w/m
·
k,较纯的环氧树脂(0.201w/m
·
k)提高了428.9%,由于氮化硼本身热导率较高,所以复合材料在30%的质量分数的氮化硼的情况下,导热性能得到较大提升。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。