光引发剂
背景技术:
1.uv固化的动态发展取决于持续创新,以支持这项技术克服持续出现的新挑战,这反映在制剂所需的新材料的快速开发上。特别地,一个关键成分是光引发剂,它的作用是将光转化为以反应中间体形式的化学能。这些中间体是能够引发存在于待固化制剂中的双键的自由基聚合的自由基。
2.光引发剂领域的发展受到多种因素的推动。第一,现有应用(如涂料、油墨、粘合剂或电子产品)的光引发剂不断改进,因为没有单一的光引发剂能够满足每种应用的所有要求,例如线速度、表面固化、固化后变色或溶解度。其次,led灯等新型灯具的推出,刺激了在该波长上调谐的光引发剂的发展。第三,越来越多的光引发剂因有毒或生殖毒性而被禁用。
3.过去几年,人们进行了各种尝试,以开发能够模仿标准光引发剂或克服诸如泛黄、线速度较高、在led灯下固化、深度固化等问题的新型光引发剂,一些例子包括3
‑
香豆素酮(us9951034,wo2017/216699),酰基锗光引发剂(ep3150641,ep2649981),苯甲酰苯基碲化物光引发剂(macromolecules,2014,47(16),5526
‑
5531),硅基光引发剂(jp2010229169,macromolecules,2009,42(16),6031
‑
6037,macromolecules 2007,40(24),8527
‑
8530,macromol.rapid commun.2017,38,1600470,macromolecules,2017,50(17),6911
–
6923)。
4.特别是,硅基类光引发剂得到了广泛的探索,但不幸的是,已知的光引发剂不能够显著改善至少一个上述提及的特性。近年来探索的第一类硅基光引发剂是α
‑
二甲硅烷氧基酮,属于α
‑
羟基酮类,但与母体化合物相比其反应性较差或相当,而且吸收波长也不受二甲硅烷氧基取代的积极影响。
5.发明目的
6.本发明的第一个目的是,提供包含α
‑
二甲硅烷氧基酮的新型可光固化的组合物,其相对于已知的含α
‑
二甲硅烷氧基芳基酮的组合物具有更高的性能。
7.本发明的另一个目的是,提供新的α
‑
二甲硅烷氧基芳基酮、它们作为光引发剂的用途和包含它们的可光固化的组合物。
8.本发明的另一个目的是,提供一种用于光固化烯键式不饱和化合物的方法以及由所述方法制造的制品。
9.发明概述
10.现在,我们惊奇地发现,与α
‑
二甲硅烷氧基酮共轭的供电子芳族基团产生了新的光引发剂,与现有技术中描述的α
‑
二甲硅烷氧基芳基酮(pi
‑
1macromolecules,2009,42(16),6031
‑
6037)相比,它们具有高反应性。
11.事实上,出乎意料地发现,被α
‑
二甲硅烷氧基芳基酮的芳基部分上的供电子基团取代,例如二苯基硫醚或对吗啉代苯基,会产生性能更好的光引发剂,其反应性是上述参考化合物(pi
‑
1)的两倍。
12.此外,我们惊讶地发现,属于α
‑
二甲硅烷氧基芳基酮类的所有化合物都可以被敏化,从而改善光谱工作波长并允许全部所述化合物在led波长(例如400nm)下工作(效果未知),而使用其他光引发剂(如α
‑
羟基酮)无法实现这一结果。
13.因此,在包含作为光引发剂的α
‑
二甲硅烷氧基芳基酮的可光固化的组合物中加入敏化剂,可以获得意想不到的技术改进,这将从以下实验实施例中显而易见。
技术实现要素:
14.根据本发明的一方面,本发明涉及一种可光固化的组合物,包括:
15.a)50至99.9重量%,优选70至98.9重量%的至少一种烯键式不饱和化合物的固体含量,不包括水或溶剂;
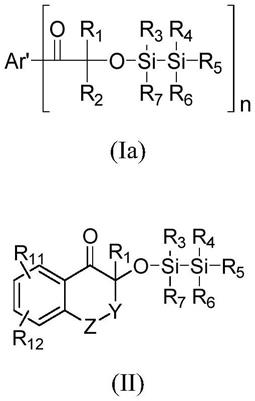
16.b)0.1至35重量%,优选0.1至20重量%,更优选0.2至15重量%的至少一种式(i)和/或(ii)化合物的固体含量,不包括水或溶剂,
[0017][0018][0019]
式中:
[0020]
‑
n是1或者2;
[0021]
‑
当n是1,ar选自以下基团:
[0022]
‑
c6‑
c
12
芳基,所述芳基是未取代的或者被一或多个选自以下组内的取代基取代:卤素,
‑
cn,
‑
cooh,
‑
oh,c1‑
c
18
烷基,
‑
o烷基,
‑
o烷氧基,
‑
o苯基,
‑
sh,
‑
s烷基,
‑
s烷氧基,
‑
s苯基,
‑
so烷基,
‑
so2烷基,
‑
so2烷氧基,
‑
so2苯基,
‑
coo烷基,so2nh2,
‑
so2nh烷基,
‑
so2n(烷基)2,
‑
nh烷基,
‑
nh烷氧基,
‑
n(烷基)2,吗啉代,哌啶子基,哌嗪子基,
‑
n(烷氧基)2,
‑
nhco烷基,
‑
nhco苯基;以及
[0023]
‑
以下基团中之一:吡啶基,噻吩基,2
‑
甲基噻吩基,吡咯基,呋喃基,茚满基,咪唑基,噻唑基,噁唑基,四氢萘基,萘基,苯并噻吩基,苯并吡咯基,苯并呋喃基,苯并咪唑基,苯并噻唑基,苯并噁唑基,3,4
‑
亚乙二氧噻吩,咔唑基,n
‑
烷基咔唑基,噻蒽基,吩噁噻基(phenoxathiinyl),吩噻嗪基,吩噁嗪基,5,10
‑
二氢吩嗪基;所有所述基团可能被一个或多个供电子基团取代;
[0024]
‑
当n是2,ar选自以下基团:c6‑
c
12
亚芳基,c6‑
c
12
亚杂芳基和
‑
亚芳基
‑
t
‑
亚芳基
‑
基团(例如
–
亚苯基
‑
t
‑
亚苯基
‑
基团)以及三甲基
‑
苯基
‑
茚满基团,例如下式所示的基团,
[0025]
[0026]
所有所述基团可能被一个或多个供电子基团取代;
[0027]
‑
y选自直接键,
‑
ch2‑
,
‑
ch2‑
ch2‑
,
‑
o
‑
,
‑
s
‑
和
‑
n烷基;
[0028]
‑
z选自直接键,
‑
o
‑
,
‑
s
‑
,
‑
so2‑
,
‑
nalk,
‑
ch2‑
和
–
c(ch3)2;
[0029]
‑
t选自直接键,
‑
o
‑
,
‑
s
‑
,
‑
so2‑
,
‑
ch2‑
,
‑
ch2ch2‑
,
‑
ch2och2‑
和
–
ch=ch
‑
;
[0030]
‑
r1选自c1‑
c8烷基,所述烷基是未取代的或者被以下取代基取代:c2‑
c8酰氧基,
‑
nr8r9,
‑
coo烷基,或者
–
cn,或者表示c3‑
c5烯基,c5‑
c
12
环烷基,苯基和苄基;
[0031]
‑
r2选自r1或者r2表示
–
ch2ch2r
10
基团;或者r1和r2与它们所结合的碳原子一起表示c5‑
c
12
环烷基,c2‑
c8‑
亚烷基或c3‑
c9氧杂
‑
或氮杂
‑
亚烷基;
[0032]
‑
r3,r4,r5,r6和r7各自独立地选自以下基团:
[0033]
‑
苯基,c1‑
c8烷基,二者是未取代的或者被以下取代基取代:
–
oh,
‑
o烷基,c2‑
c8酰氧基,
‑
nr8r9,
‑
coo烷基或者cn;以及
[0034]
‑
o烷基;
[0035]
‑
r8和r9各自独立地选自以下基团:c1‑
c
18
烷基,所述烷基是未取代的或者被以下取代基取代:
–
oh,
‑
o烷基,或者
–
cn;
[0036]
‑
r
10
选自以下基团:
‑
conh2,
‑
conh烷基,
‑
con(烷基)2和
‑
p(=o)(o烷基)2[0037]
‑
r
11
选自以下基团:氢,卤素,
‑
cn,
‑
cooh,
‑
oh,c1
‑
c18烷基,
‑
o烷基,
‑
o烷氧基,
‑
o苯基,
‑
sh,
‑
s烷基,
‑
s烷氧基,
‑
s苯基,
‑
soalk,
‑
so2烷基,
‑
so2烷氧基,
‑
so2苯基,
‑
coo烷基,so2nh2,
‑
so2nh烷基,
‑
so2n(烷基)2,
‑
nh烷基,
‑
nh烷氧基,
‑
n(烷基)2,吗啉代,哌啶子基,哌嗪子基,
‑
n(烷氧基)2,
‑
nhco烷基以及
–
nhco苯基;
[0038]
‑
r
12
具有如r
11
的含义之一;
[0039]
‑
或者r
11
和r
12
与它们所结合的碳原子一起表示c5‑
c
12
环烷基或c6‑
c
12
芳基;
[0040]
c)0.01至15重量%,优选0.01至10重量%,更优选0.02至8重量%的至少一种光敏剂的固体含量,不包括水或溶剂,所述光敏剂来自具有三重态能量225
‑
310kj/mol的芳香族羰基化合物。
[0041]
根据本发明的另一方面,本发明涉及如式(ia)和(ii)所示的光引发剂:
[0042][0043][0044]
式中:
[0045]
‑
n是1或者2;
[0046]
‑
当n是1,ar选自以下基团之一:c6‑
c
12
芳基,噻吩基,2
‑
甲基噻吩基,呋喃基,茚满基,噻唑基,噁唑基,四氢萘基,苯并噻吩基,苯并吡咯基,苯并呋喃基,咔唑基,n
‑
烷基咔唑
基,噻蒽基,吩噁噻基,吩噻嗪基,吩噁嗪基和5,10
‑
二氢吩嗪基;所有所述基团可能被一个或多个供电子基团取代;条件是当c6‑
c
12
芳基苯基时,则所述苯基总是被一个或多个供电子基团取代;
[0047]
‑
当n是2,ar选自以下基团:c6‑
c
12
亚芳基,c6‑
c
12
亚杂芳基,
–
亚芳基
‑
t
‑
亚芳基
‑
基团(例如
–
亚苯基
‑
t
‑
亚苯基
‑
基团)和三甲基
‑
苯基
‑
茚满基团,例如下式所示的基团,
[0048][0049]
所有所述基团可能被一个或多个供电子基团取代;
[0050]
‑
y选自直接键,
‑
ch2‑
,
‑
ch2‑
ch2‑
,
‑
o
‑
,
‑
s
‑
和
‑
n烷基;
[0051]
‑
z选自直接键,
‑
o
‑
,
‑
s
‑
,
‑
so2‑
,
‑
nalk,
‑
ch2‑
和
‑
c(ch3)2;
[0052]
‑
t选自直接键,
‑
o
‑
,
‑
s
‑
,
‑
so2‑
,
‑
ch2‑
,
‑
ch2ch2‑
,
‑
ch2och2‑
和
–
ch=ch
‑
;
[0053]
‑
r1选自氢,c1‑
c8烷基,所述烷基是未取代的或者被以下取代基取代:c2‑
c8酰氧基,
‑
nr8r9,
‑
coo烷基,或者
–
cn,或者r1表示c3‑
c5烯基,c5‑
c
12
环烷基,苯基和苄基;
[0054]
‑
r2选自r1或者r2表示
–
ch2ch2r
10
基;或者r1和r2与它们所结合的碳原子一起表示c5‑
c
12
环烷基,c2‑
c8‑
亚烷基或c3‑
c9氧杂
‑
或氮杂
‑
亚烷基;
[0055]
‑
r3,r4,r5,r6和r7各自独立地选自以下基团:
[0056]
‑
苯基,c1‑
c8烷基,二者是未取代的或者被以下取代基取代:
–
oh,
‑
o烷基,c2‑
c8酰氧基,
‑
nr8r9,
‑
coo烷基或者cn;以及
[0057]
‑
o烷基;
[0058]
‑
r8和r9各自独立地选自以下基团:c1‑
c
18
烷基,所述烷基是未取代的或者被以下取代基取代:
–
oh,
‑
o烷基,或者
–
cn;
[0059]
‑
r
10
选自以下基团:
‑
conh2,
‑
conh烷基,
‑
con(烷基)2和
‑
p(=o)(o烷基)2;
[0060]
‑
r
11
选自以下基团:氢,卤素,
‑
cn,
‑
cooh,
‑
oh,c1
‑
c18烷基,
‑
o烷基,
‑
o烷氧基,
‑
o苯基,
‑
sh,
‑
s烷基,
‑
s烷氧基,
‑
s苯基,
‑
soalk,
‑
so2烷基,
‑
so2烷氧基,
‑
so2苯基,
‑
coo烷基,so2nh2,
‑
so2nh烷基,
‑
so2n(烷基)2,
‑
nh烷基,
‑
nh烷氧基,
‑
n(烷基)2,吗啉代,哌啶子基,哌嗪子基,
‑
n(烷氧基)2,
‑
nhco烷基和
–
nhco苯基;
[0061]
‑
r
12
具有如r
11
的含义之一;
[0062]
‑
或者r
11
和r
12
与它们所结合的碳原子一起表示c5‑
c
12
环烷基或c6‑
c
12
芳基;并且其中:
[0063]
‑
当r1和r2与它们所结合的碳原子一起表示c5‑
c
12
环烷基,c2‑
c8‑
亚烷基或c3‑
c9氧杂
‑
或氮杂
‑
亚烷基时,则ar'也可以不被取代,优选是c6‑
c
10
环烷基,更优选环己基。
[0064]
根据本发明的再一方面,本发明涉及一种可光固化的组合物,包括:
[0065]
a)50至99.9重量%,优选70至98.9重量%的至少一种烯键式不饱和化合物的固体含量,不包括水或溶剂;
[0066]
b)0.1至35重量%,优选0.1至20重量%,更优选0.2至15重量%的至少一种如上所定义的式(ia)和/或(ii)化合物的固体含量,不包括水或溶剂。
[0067]
根据本发明的另一方面,本发明涉及如上定义的式(ia)和(ii)化合物作为光引发剂的用途。
[0068]
根据本发明的另一方面,本发明涉及一种光固化组合物方法,该组合物包括式(i)、(ia)和(ii)化合物中的至少一种,以及涉及由所述方法制造的制品。
[0069]
根据本发明的另一方面,本发明涉及使用敏化剂来加宽二甲硅烷基芳基酮的光谱工作波长的用途。
具体实施方式
[0070]
根据本发明,术语“光固化”和“光聚合”(及其衍生术语)是可互换的。
[0071]“α
‑
二甲硅烷氧基芳基酮”是指以式(i)、(ia)和(ii)所示的本发明化合物,在本文中包括如所述式定义的α
‑
二甲硅烷氧基芳基酮和α
‑
二甲硅烷氧基杂芳基酮。
[0072]
除非另有说明,术语“取代的苯基”是指被一个或多个供电子基团取代的苯基。
[0073]
‑
优选地,所述供电子基团选自:oh、c1‑
c
18
烷基、
‑
o烷基、
‑
o烷氧基、
‑
o苯基、
‑
sh、
‑
s烷基、
‑
s烷氧基、
‑
s苯基、
‑
so2烷基、
‑
so2烷氧基、
‑
so2苯基、
‑
coo烷基、so2nh2、
‑
so2nh烷基、
‑
so2n(烷基)2、
‑
nh烷基、
‑
nh烷氧基、
‑
n(烷基)2、吗啉代、哌啶子基、哌嗪子基、
‑
n(烷氧基)2、
‑
nhco烷基或
‑
nhco苯基、或者代表噻吩基、2
‑
甲基噻吩基、吡咯基、呋喃基、茚满基、咪唑基、噻唑基、噁唑基、四氢萘基、萘基、苯并噻吩基、苯并吡咯基、苯并呋喃基、苯并咪唑基、苯并噻唑基、苯并噁唑基、3,4
‑
亚乙二氧噻吩、均三甲苯基(mesityl,即2,4,6
‑
三甲基苯基)、异丙基苯基、苯氧基苯基、对壬基苯基、c
10
‑
c
13
烷基苯基、羟基苯基、甲苯基、叔丁基苯基、二甲苯基、甲氧基苯基、乙氧基苯基、苯氧基苯基、甲硫基苯基、苯硫苯基、4
‑
甲氧基苯甲硫醚、丁基磺基苯基、苯基磺基苯基、乙氧基羰基苯基、叔丁氧基羰基苯、甲氨基磺苯基、二丙氨基磺苯基、二甲氨基苯基、4
‑
吗啉代苯基、苯甲酰氨基苯基和乙酰氨基苯基。
[0074]
根据一个优选的实施方案,在本发明的式(i)、(ia)和(ii)中:
[0075]
‑
当n为1时,ar选自以下基团:苯基、氟苯基、溴苯基、氯苯基、二氯苯基、碘苯基、氰基苯基、萘基、菲基、蒽基、二苯基、均三甲苯、异丙基苯基、异丙基氯苯基、苯氧基苯基、对壬基苯基、c
10
‑
c
13
烷基苯基、羟苯基、甲苯基、氯甲苯基、叔丁基苯基、二甲苯基、溴二甲苯基、甲氧基苯基、乙氧基苯基、苯氧基苯基、甲基噻吩基、苯基噻吩基、4
‑
甲氧基苯甲硫醚、丁基磺苯基、苯基磺苯基、乙氧基羰基苯基、叔丁氧基羰基苯基、甲基氨基磺苯基、二丙基氨基磺苯基、二甲氨基苯基、4
‑
吗啉代苯基、苯甲酰氨基苯基、乙酰基氨基苯基、吡啶基噻吩基、2
‑
甲基噻吩基、呋喃基、茚满基、噻唑基、噁唑基、四氢萘基、苯并噻吩基、苯并吡咯基、苯并呋喃基、咔唑基、n
‑
烷基咔唑基、噻蒽基、吩噁噻基、吩噻嗪基、吩噁嗪基和5,10
‑
二氢吩嗪基;
[0076]
‑
当n为1时,ar’选自以下基团:
[0077]
·
萘基、菲基、蒽基、二苯基;
[0078]
·
被一或多个供电子基团取代的苯基,所述供电子基团优选选自以下基团的一或多个:oh、c1
‑
c18烷基、
‑
o烷基、
‑
o烷氧基、
‑
o苯基、
‑
sh、
‑
s烷基、
‑
s烷氧基、
‑
s苯基、
‑
so2烷基、
‑
so2烷氧基、
‑
so2苯基、
‑
coo烷基、so2nh2、
‑
so2nh烷基、
‑
so2n(烷基)2、
‑
nh烷基、
‑
nh烷氧基、
‑
n(烷基)2、吗啉代、哌啶子基、哌嗪子基、
‑
n(烷氧基)2、
‑
nhco烷基或
‑
nhco苯基,或者代表噻吩基、2
‑
甲基噻吩基、吡咯基、呋喃基、茚满基、咪唑基、噻唑基、噁唑基、四氢萘基、萘基、苯并噻吩基、苯并吡咯基、苯并呋喃基、苯并咪唑基、苯并噻唑基、苯并噁唑基、3,4
‑
亚乙
二氧基噻吩;
[0079]
·
被一或多个供电子基团取代的其他优选的苯基,选自均三甲苯基、异丙基苯基、苯氧基苯基、对壬基苯基、c
10
‑
c
13
烷基苯基、羟基苯基、甲苯基、叔丁基苯基、二甲苯基、甲氧基苯基、乙氧基苯基、苯氧基苯基、甲硫基苯基、苯硫基苯基、4
‑
甲氧基苯甲硫醚、丁基磺基苯基、苯基磺基苯基、乙氧基羰基苯基、叔丁氧基羰基苯基、甲氨基磺苯基、二丙氨基磺苯基、二甲氨基苯基、4
‑
吗啉代苯基、苯甲酰氨基苯基、乙酰氨基苯基;
[0080]
‑
c6‑
c
12
亚芳基选自亚苯基、亚萘基和二亚苯基。
[0081]
‑
c6‑
c
12
亚杂芳基选自未取代的或取代的苯并呋喃、咔唑和香豆素酮基团;
[0082]
‑
亚芳基
‑
t
‑
亚芳基优选选自亚苯基
‑
t
‑
亚苯基,例如二苯甲烷、二苯醚和二苯硫醚;及三甲基
‑
苯基
‑
茚满基团,如下式所示的基团:
[0083][0084]
‑
c1‑
c
18
烷基选自直链或支链的饱和烷基,其可以是未取代的或被芳基、烷基、芳氧基、烷氧基、杂原子和/或杂环基取代,包括例如甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基、戊基、己基、庚基、辛基、2
‑
乙基己基、2,4,4
‑
三甲基戊基、癸基、十二烷基、十四烷基、1,1
‑
二甲基丙基、1,1
‑
二甲基丁基、1,1,3,3
‑
四甲基丁基、苄基、1
‑
苯基乙基、2
‑
苯基
‑
乙基、α,α
‑
二甲基苄基、二苯甲基、对甲苯基甲基、1
‑
(对丁基苯基)乙基、对氯苄基、2,4
‑
二氯苄基、对甲氧基苄基、间乙氧基苄基、2
‑
羟基乙基、2
‑
羟基丙基、3
‑
羟基丙基、4
‑
羟基丁基、6
‑
羟基己基、2
‑
甲氧基乙基、2
‑
甲氧基丙基、3
‑
甲氧基丙基、4
‑
甲氧基丁基、6
‑
甲氧基己基、2
‑
乙氧基乙基、2
‑
乙氧基丙基、3
‑
乙氧基丙基、4
‑
乙氧基丁基或6
‑
乙氧基己基、2
‑
甲氧基羰基乙基、2
‑
乙氧基羰基乙基、2
‑
丁氧基羰基丙基、1,2
‑
二(甲氧基羰基)乙基、2
‑
丁氧基乙基、二乙氧基甲基、二乙氧基乙基、1,3
‑
二氧戊环
‑2‑
基、1,3
‑
二氧杂环己烷
‑2‑
基、2
‑
甲基
‑
1,3
‑
二氧戊环
‑2‑
基、4
‑
甲基
‑
1,3
‑
二氧戊环
‑2‑
基、2
‑
异丙氧基乙基、2
‑
丁氧基丙基、2
‑
辛氧基乙基、氯甲基、2
‑
氯乙基、三氯甲基、三氟甲基、1,1
‑
二甲基
‑2‑
氯乙基、2
‑
甲氧基异丙基、丁硫基甲基、2
‑
十二烷基硫乙基、2
‑
苯硫基乙基、2,2,2
‑
三氟乙基、2
‑
氨基乙基、2
‑
氨基丙基、3
‑
氨基丙基、4
‑
氨基丁基、6
‑
氨基己基、2
‑
甲基氨基乙基、2
‑
甲基氨基丙基、3
‑
甲基氨基丙基、4
‑
甲基氨基丁基、6
‑
甲基氨基己基、2
‑
二甲基氨基乙基、2
‑
二甲基氨基丙基、3
‑
二甲基氨基丙基、4
‑
二甲基氨基丁基、6
‑
二甲基氨基己基、2
‑
羟基
‑
2,2
‑
二甲基乙基、2
‑
苯氧基乙基、2
‑
苯氧基丙基、3
‑
苯氧基丙基、4
‑
苯氧基丁基、以及6
‑
苯氧基己基;
[0085]
‑
c1‑
c8烷基选自直链或支链的、饱和的、未取代的烷基,例如甲基、乙基、丙基、异丙基、正丁基、仲丁基或叔丁基、异戊基、正己基、2
‑
乙基己基或辛基;以及
[0086]
‑
c5‑
c
12
环烷基可以是未取代的或被芳基、烷基、芳氧基、烷氧基、杂原子和/或杂环基取代的基团,包括例如环戊基、环己基、环辛基、环十二烷基、甲基环戊基、2,5
‑
二甲基环戊基、甲基环己基、2,6
‑
二甲基环己基、2,6
‑
二乙基环己基、丁基环己基、甲氧基环己基、2,6
‑
二甲氧基环己基、2,6
‑
二乙氧基环己基、丁基硫基环己基、氯代环己基、2,6
‑
二氯环己基和2,5
‑
二氯环戊基;或者c5‑
c
12
环烷基可以是饱和或不饱和的、未取代的或取代的双环系统
(例如降冰片基或降冰片烯基)、三环系统(例如金刚烷基)。
[0087]
除非明确指出,烷基和烷氧基是c1‑
c6直链或支链的饱和烷基或烷氧基,例如c1‑
c4基团。
[0088]
优选地,n为1。
[0089]
优选地,ar选自苯基或取代的苯基,例如c
10
‑
c
13
烷基苯基、甲苯基、叔丁基苯基、甲氧基苯基、乙氧基苯基、苯氧基苯基、甲硫基苯基、苯硫基苯基;4
‑
甲氧基苯甲硫醚;二甲氨基苯基;4
‑
吗啉代苯基;噻吩基;2
‑
甲基噻吩基;茚满基;噻唑基;噁唑基;四氢萘基;苯并噻吩基;苯并吡咯基和苯并呋喃基。
[0090]
优选地,ar’是具有一个或多个供电子基团的取代苯基,例如c
10
‑
c
13
烷基苯基、甲苯基、叔丁基苯基、甲氧基苯基、乙氧基苯基、苯氧基苯基、甲硫基苯基、苯硫基苯基、4
‑
甲氧基苯甲硫醚、二甲氨基苯基、4
‑
吗啉代苯基。
[0091]
优选地,ar’是噻吩基、2
‑
甲基噻吩基、茚满基、噻唑基、噁唑基、四氢萘基、苯并噻吩基、苯并吡咯基或苯并呋喃基。
[0092]
优选地,r1和r2中至少一个不是氢,更优选r1和r2都不是氢。
[0093]
优选地,r1是甲基、乙基、丙基或苄基。
[0094]
优选地,r2是甲基、乙基或丙基,或者r2与r1和它们所键合的碳原子一起形成c5‑
c
12
环烷基。
[0095]
优选地,ar’是未取代的并且r1和r2以及它们所结合的碳原子形成c5‑
c
12
环烷基,优选c6‑
c
10
环烷基,更优选环己基。
[0096]
优选地,r3,r4,r5,r6,r7各自独立地为甲基、乙基或苯基。
[0097]
优选地,r
11
和r
12
是氢。
[0098]
式(i)、(ia)和(ii)所示的优选的α
‑
二甲硅烷氧基芳基酮列于表1中,仅用于说明目的,但不限于此。
[0099]
表1
[0100]
[0101]
[0102]
[0103]
[0104][0105]
式(i)、(ia)和(ii)的化合物能够根据本领域技术人员已知的常规方法制备,例如根据以下流程从相应的α
‑
羟基酮进行甲硅烷基化反应而制备:
[0106][0107]
式(i)、(ia)和(ii)的代表性化合物的示意性合成报道于下文的实例中。
[0108]
根据本发明,式(i)、(ia)和(ii)的光引发剂能够用于制备包含烯键式不饱和化合物a)的可光固化的组合物。
[0109]
不饱和化合物a)可以含有一个或多个烯族双键。它们可以具有低分子量(单体)或高分子量(低聚)。
[0110]
具有一个双键的合适的低分子量单体的实例是烷基或羟基烷基丙烯酸酯或甲基丙烯酸酯,例如甲基、乙基、丁基、2
‑
乙基己基或2
‑
羟基乙基丙烯酸酯,丙烯酸异冰片酯和甲基或乙基甲基丙烯酸酯。同样令人感兴趣的是硅或氟改性的树脂,例如硅氧烷丙烯酸酯。这些单体的其它实例是丙烯腈、丙烯酰胺、甲基丙烯酰胺、n
‑
取代的(甲基)丙烯酰胺、苯乙烯、烷基苯乙烯和卤代苯乙烯、乙烯基酯诸如乙酸乙烯酯、乙烯基醚诸如异丁基乙烯基醚、n
‑
乙烯基吡咯烷酮、氯乙烯或偏二氯乙烯。
[0111]
具有一个以上双键的单体的实例是二丙烯酸乙二醇酯、二丙烯酸丙二醇酯、二丙烯酸新戊二醇酯、二丙烯酸六亚甲基二醇酯和双酚a二丙烯酸酯、4,4'
‑
双(2
‑
丙烯酰氧基乙氧基)
‑
二苯基丙烷、三羟甲基丙烷三丙烯酸酯、三丙烯酸季戊四醇酯或四丙烯酸季戊四醇酯、丙烯酸乙烯酯、二乙烯基苯、琥珀酸二乙烯酯、邻苯二甲酸二烯丙酯、磷酸三烯丙酯、异氰脲酸三烯丙酯和三(2
‑
丙烯酰基乙基)异氰脲酸酯。
[0112]
高分子量(低聚)多不饱和化合物的实例是丙烯酸酯化环氧树脂、丙烯酸酯化或含乙烯基醚或环氧基的聚酯、丙烯酸酯化聚氨酯或丙烯酸酯化聚醚。不饱和低聚物的其它实例是不饱和聚酯树脂,其通常由马来酸、邻苯二甲酸和一种或多种二醇制备并且分子量为约500至3000da。这种不饱和低聚物也可以称为预聚物。
[0113]
特别适合于实施本发明的化合物a)的实例是烯属不饱和羧酸和多元醇或聚环氧化物的酯,以及在主链中或在侧基中含有烯属不饱和基团的聚合物,例如不饱和聚酯、聚酰胺和聚氨酯及其共聚物、醇酸树脂、聚丁二烯和丁二烯共聚物、聚异戊二烯和异戊二烯共聚物、在侧链具有(甲基)丙烯酸基团的聚合物和共聚物、以及一种以上这样的聚合物的混合物。
[0114]
用于制备所述酯的不饱和羧酸或酸酐的示意性实例是丙烯酸、甲基丙烯酸、马来酸酐、丁烯酸、衣康酸、肉桂酸以及不饱和脂肪酸如亚麻酸和油酸。优选的是丙烯酸和甲基丙烯酸。
[0115]
可以酯化的多元醇是芳族、脂族和脂环族多元醇,优选为脂族和脂环族多元醇。
[0116]
芳族多元醇例如是氢醌、4,4'
‑
二羟基二苯基、2,2
‑
二(4
‑
羟基苯基)丙烷以及酚醛清漆(novolak)和甲阶酚醛树脂(resole)。可以酯化的聚环氧化物包括基于所述多元醇,特别是芳族多元醇和环氧氯丙烷的那些。同样适合作为多元醇的是在聚合物链或侧基中含有羟基的聚合物和共聚物,例如聚乙烯醇及其共聚物、或聚甲基丙烯酸羟基烷基酯或其共聚物。其它合适的多元醇是带有羟基端基的低聚酯。
[0117]
脂族和脂环族多元醇的实例包括优选含有2至12个碳原子的亚烷基二醇,例如乙二醇、1,2
‑
或1,3
‑
丙二醇、1,2
‑
、1,3
‑
或1,4
‑
丁二醇、戊二醇、己二醇、辛二醇、十二烷二醇、二乙二醇、三乙二醇、分子量优选为200
‑
1500da的聚乙二醇、1,3
‑
环戊二醇、1,2
‑
、1,3
‑
或1,4
‑
环己二醇、1,4
‑
二羟基甲基环己烷、甘油、三(β
‑
羟基乙基)胺、三羟甲基乙烷、三羟甲基丙烷、季戊四醇、二季戊四醇和山梨醇。
[0118]
其它合适的烯属不饱和化合物a)是由不饱和羧酸和优选具有2至6个,更优选2至4个氨基的芳族、脂族和脂环族聚胺获得的不饱和聚酰胺。这种聚胺的实例是:乙基烯二胺、1,2
‑
或1,3
‑
丙基烯二胺、1,2
‑
、1,3
‑
或1,4
‑
丁二胺、1,5
‑
戊二胺、1,6
‑
己二胺、辛二胺、十二
亚甲基二胺、1,4
‑
二氨基环己烷、异佛尔酮二胺、苯二胺、双苯二胺(双苯二胺)、二(β
‑
氨基乙基)醚、二亚乙基三胺、三亚乙基四胺和二(β
‑
氨基乙氧基)
‑
和二(β
‑
氨基丙氧基)乙烷。其它合适的聚胺是可以在侧链中含有额外氨基的聚合物和共聚物,以及含有氨基端基的低聚酰胺。
[0119]
这种不饱和聚酰胺的具体实例是:亚甲基双丙稀酰胺、1,6
‑
六亚甲基双丙烯酰胺、二亚乙基三胺三甲基丙烯酰胺、双(甲基丙烯酰胺基丙氧基)乙烷和n
‑
[(β
‑
羟基乙氧基)乙基]
‑
丙烯酰胺。
[0120]
不饱和聚氨酯也适合实施本发明,例如衍生自例如饱和或不饱和二异氰酸酯以及不饱和或饱和二醇的那些。聚丁二烯和聚异戊二烯及其共聚物也是有用的。合适的共聚单体包括例如烯烃(诸如乙烯、丙烯、丁烯和己烯)、(甲基)丙烯酸酯、丙烯腈、苯乙烯和氯乙烯。
[0121]
在侧链中具有不饱和(甲基)丙烯酸酯基团的聚合物可以用作组分a)。典型地,它们可以是基于酚醛清漆的环氧树脂与(甲基)丙烯酸的反应产物;已用(甲基)丙烯酸酯化的乙烯醇或其羟基烷基衍生物的均聚物或共聚物;和已用羟基烷基(甲基)丙烯酸酯酯化的(甲基)丙烯酸酯的均聚物和共聚物。
[0122]
混合物中的组分c)是三重态能量为225
‑
310kj/mol的光敏剂。
[0123]
光敏剂的实例是本领域技术人员常用的那些,芳族羰基化合物,例如二苯甲酮、噻吨酮、蒽醌和3
‑
酰基香豆素衍生物、三联苯、苯乙烯基酮和3
‑
(芳酰基亚甲基)
‑
噻唑啉、樟脑醌以及曙红、若丹明和赤藓红染料。
[0124]
噻吨酮的实例是噻吨酮、2
‑
异丙基噻吨酮、2
‑
氯噻吨酮、2
‑
十二烷基噻吨酮、2,4
‑
二乙基噻吨酮、2,4
‑
二甲基噻吨酮、1
‑
甲氧基羰基噻吨酮、2
‑
乙氧基羰基噻吨酮、3
‑
(2
‑
甲氧基乙氧基羰基)噻吨酮、4
‑
丁氧基羰基噻吨酮、3
‑
丁氧基羰基
‑7‑
甲基噻吨酮、1
‑
氰基
‑3‑
氯噻吨酮、1
‑
乙氧基羰基
‑3‑
氯噻吨酮、1
‑
乙氧基羰基
‑3‑
乙氧基噻吨酮、1
‑
乙氧基羰基
‑3‑
氨基噻吨酮、1
‑
乙氧基羰基
‑3‑
苯基磺酰基噻吨酮、3,4
‑
二[2
‑
(2
‑
甲氧基乙氧基)乙氧基羰基]噻吨酮、1
‑
乙氧基羰基
‑3‑
(1
‑
甲基
‑1‑
吗啉代乙基)噻吨酮、2
‑
甲基
‑6‑
二甲氧基甲基噻吨酮、2
‑
甲基
‑6‑
(1,1
‑
二甲氧基芐基)噻吨酮、2
‑
吗啉代甲基噻吨酮、2
‑
甲基
‑6‑
吗啉代甲基噻吨酮、n
‑
烯丙基噻吨酮
‑
3,4
‑
二甲酰亚胺、n
‑
辛基噻吨酮
‑
3,4
‑
二甲酰亚胺、n
‑
(1,1,3,3
‑
四甲基丁基)
‑
噻吨酮
‑
3,4
‑
二甲酰亚胺、1
‑
苯氧基噻吨酮、6
‑
乙氧基羰基
‑1‑2‑
甲氧基噻吨酮、6
‑
乙氧基羰基
‑2‑
甲基噻吨酮,噻吨酮
‑2‑
聚乙二醇酯、2
‑
羟基
‑3‑
(3,4
‑
二甲基
‑9‑
氧代
‑
9h
‑
噻吨酮
‑2‑
基氧基)
‑
n,n,n
‑
三甲基
‑1‑
氯化丙铵,或者是专利申请pct/ep2011/069514描述的那些,例如正十二烷基
‑7‑
甲基
‑
噻吨酮
‑3‑
羧酸酯以及n,n
‑
二异丁基
‑7‑
甲基
‑
噻吨酮
‑3‑
脲。同样合适的是聚合物噻吨酮衍生物(例如来自igm resins b.v.的omnipol tx、来自rahn a.g.的genopol tx
‑
1、来自lambson limited的speedcure 7010)。
[0125]
二苯甲酮的实例是二苯甲酮、4
‑
苯基二苯甲酮、4
‑
甲氧基二苯甲酮、4,4'
‑
二甲氧基二苯甲酮、4,4'
‑
二甲基二苯甲酮、4,4'
‑
二氯二苯甲酮、4,4'
‑
二甲基氨基二苯甲酮、4,4'
‑
二乙基氨基二苯甲酮、4
‑
甲基二苯甲酮、2,4,6
‑
三甲基二苯甲酮、4
‑
(4
‑
甲基硫代苯基)二苯甲酮、3,3'
‑
二甲基
‑4‑
甲氧基二苯甲酮、2
‑
苯甲酰基苯甲酸甲酯、4
‑
(2
‑
羟基乙基硫代)二苯甲酮、4
‑
(4
‑
甲苯基硫代)二苯甲酮、4
‑
苯甲酰基
‑
n,n,n
‑
三甲基苯甲基氯化铵、2
‑
羟基
‑3‑
(4
‑
苯甲酰基苯氧基)
‑
n,n,n
‑
三甲基
‑1‑
丙基氯化铵一水合物、4
‑
(13
‑
丙烯酰基
‑
1,4,7,
10,13
‑
五氧杂十三烷基)二苯甲酮、4
‑
苯甲酰基
‑
n,n
‑
二甲基
‑
n
‑
[2
‑
(1
‑
氧代
‑2‑
丙烯基)氧基乙基
‑
苯甲基氯化铵。同样合适的是聚合物二苯甲酮衍生物(例如来自igm resins b.v.的bp、2702和682、来自rahn a.g.的bp
‑
2以及来自lambson limited的7005)。
[0126]3‑
酰基香豆素衍生物的实例是3
‑
苯甲酰基香豆素、3
‑
苯甲酰基
‑7‑
甲氧基香豆素、3
‑
苯甲酰基
‑
5,7
‑
二(丙氧基)香豆素、3
‑
苯甲酰基
‑
6,8
‑
二氯香豆素、3
‑
苯甲酰基
‑6‑
氯香豆素、3,3'
‑
羰基
‑
双[5,7
‑
二(丙氧基)香豆素]、3,3'
‑
羰基
‑
双(7
‑
甲氧基香豆素)、3,3'
‑
羰基
‑
双(7
‑
二乙基氨基香豆素)、3
‑
异丁酰基香豆素、3
‑
苯甲酰基
‑
5,7
‑
二甲氧基香豆素、3
‑
苯甲酰基
‑
5,7
‑
二乙氧基香豆素、3
‑
苯甲酰基
‑
5,7
‑
二丁氧基香豆素、3
‑
苯甲酰基
‑
5,7
‑
二(甲氧基乙氧基)香豆素、3
‑
苯甲酰基
‑
5,7
‑
二(烯丙基氧基)香豆素、3
‑
苯甲酰基
‑7‑
二甲基氨基香豆素、3
‑
苯甲酰基
‑7‑
二乙基氨基香豆素、3
‑
异丁酰基
‑
1,7
‑
二甲基氨基香豆素、5,7
‑
二甲氧基
‑3‑
(1
‑
萘甲酰基)香豆素、5,7
‑
二甲氧基
‑
3(1
‑
萘甲酰基)
‑
香豆素、3
‑
苯甲酰基苯并[f]香豆素、7
‑
二乙基氨基
‑3‑
噻吩酰基香豆素、3
‑
(4
‑
氰基苯甲酰基)
‑
5,7
‑
二甲氧基香豆素,或者ep2909243和wo2017/216699中描述的那些。
[0127]3‑
(芳酰基亚甲基)
‑
噻唑啉的实例是3
‑
甲基
‑
1,2
‑
苯甲酰基亚甲基
‑
β
‑
萘并噻唑啉、3
‑
甲基
‑2‑
苯甲酰基亚甲基
‑
苯并噻唑啉、3
‑
乙基
‑2‑
丙酰基亚甲基
‑
β
‑
萘并噻唑啉。
[0128]
其它芳香族羰基化合物的实例是苯乙酮、3
‑
甲氧基苯乙酮、4
‑
苯基苯乙酮、苯偶酰、wo 2013/164394中描述的那些、2
‑
乙酰基萘、2
‑
萘甲醛、9,10
‑
蒽醌、9
‑
芴酮、二苯并环庚酮、呫吨酮、2,5
‑
双(4
‑
二乙基氨基亚芐基)环戊酮、α
‑
(对二甲基氨基亚芐基);酮类,例如2
‑
(4
‑
二甲基氨基
‑
亚芐基)
‑
茚
‑1‑
酮或3
‑
(4
‑
二甲基氨基苯基)
‑1‑
茚
‑5‑
基
‑
丙烯酮、3
‑
苯基硫代邻苯二甲酰亚胺、n
‑
甲基
‑
3,5
‑
二(乙基硫代)邻苯二甲酰亚胺。
[0129]
特别优选的是噻吨酮和3
‑
酰基香豆素。
[0130]
我们惊奇地发现,所述组分c)增加了光引发剂b)的活性而不会缩短混合物的保存期。此外,混合物的一个特殊优点是,选择合适的光敏剂c)可使光引发剂b)的光谱灵敏度转移到任何所希望的波长区域。
[0131]
除了组分a)、b)和c)或者a)和d)之外,本发明的可光固化的组合物还可包含其他光引发剂e)和/或添加剂f)。
[0132]
其他光引发剂e)的含量可以占固体含量(不包括水或溶剂)的0.5
‑
15重量%,优选占组合物的1
‑
10重量%。
[0133]
合适的其他光引发剂e)的实例是樟脑醌,二苯甲酮,二苯甲酮衍生物,苯乙酮,苯乙酮衍生物,二烷氧基苯乙酮,α
‑
羟基酮,α
‑
氨基酮,4
‑
芳酰基
‑
1,3
‑
二氧戊环;安息香烷基醚和苯缩酮,例如苯偶酰二甲基缩酮;酮砜类,例如1
‑
[4
‑
[(4
‑
苯甲酰基
‑
苯基)
‑
硫代]
‑
苯基]
‑2‑
甲基
‑2‑
[(4
‑
甲基
‑
苯基)
‑
磺酰基]
‑
丙烷
‑1‑
酮(1001,来自igm resins bv);3
‑
酮香豆素,例如在ep2909243和wo2017216699中描述的那些;苯基乙醛酸酯及其衍生物;二聚苯基乙醛酸酯;过酸酯类,例如二苯甲酮四羧酸过酸酯,例如在ep 126 541中描述的那些;酰基膦光引发剂(可选自单酰基氧化膦、双酰基氧化膦、三酰基氧化膦和多功能单或双酰基氧化膦);卤代甲基三嗪、六芳基双咪唑/共引发剂系统,例如邻氯六苯基双咪唑与2
‑
巯基苯并噻唑组合;二茂铁化合物或二茂钛,例如二环戊二烯基
‑
双(2,6
‑
二氟
‑3‑
吡咯并
‑
苯基)钛;o
‑
酰肟酯光引发剂。
[0134]
α
‑
羟基酮和α
‑
氨基酮的实例是1
‑
羟基环己基苯基酮、2
‑
羟基
‑2‑
甲基
‑1‑
苯基
‑
丙烷
‑1‑
酮、1
‑
[4
‑
(2
‑
羟基乙氧基)苯基]
‑2‑
羟基
‑2‑
甲基
‑1‑
丙烷
‑1‑
酮、2
‑
羟基
‑1‑
{4
‑
[4
‑
(2
‑
羟基
‑2‑
甲基
‑
丙酰基)
‑
苯甲基]苯基}
‑2‑
甲基
‑
丙烷
‑1‑
酮、2
‑
羟基
‑1‑
{4
‑
[4
‑
(2
‑
羟基
‑2‑
甲基
‑
丙酰基)
‑
苯氧基]
‑
苯基}
‑2‑
甲基
‑
丙
‑1‑
酮、2
‑
甲基
‑1‑
(4
‑
甲基硫苯基)
‑2‑
吗啉代丙烷
‑1‑
酮),2
‑
苯甲基
‑2‑
二甲基氨基
‑1‑
(4
‑
吗啉代苯基)
‑
丁烷
‑1‑
酮以及(2
‑
(二甲基氨基)
‑2‑
[(4
‑
甲基苯基)甲基]
‑1‑
[4
‑
(4
‑
吗啉基)苯基]
‑1‑
丁酮)。
[0135]
o
‑
酰肟酯光引发剂的实例是1,2
‑
辛二酮,1
‑
[4
‑
(苯硫基)苯基]
‑2‑
(o
‑
苯甲酰肟),乙酮1
‑
[9
‑
乙基
‑6‑
(2
‑
甲基苯甲酰基)
‑
9h
‑
咔唑
‑3‑
基]1
‑
(o
‑
乙酰肟)或gb2339571中描述的那些。
[0136]
酰基膦光引发剂的实例包括但不限于:双(2,4,6
‑
三甲基苯甲酰基)苯基氧化膦,双(2,6
‑
二甲氧基苯甲酰基)
‑
2,4,4
‑
三甲基戊基氧化膦,双(2,4,6
‑
三甲基苯甲酰基)
‑
(2,4
‑
二戊基氧基苯基),2,4,6
‑
三甲基苯甲酰基
‑
二苯基氧化膦和(2,4,6
‑
三甲基苯甲酰基)苯基次膦酸乙酯,苯基(2,4,6
‑
三甲基苯甲酰基)次膦酸、甘油乙氧基化三酯(igm resins bv的tp)。
[0137]
基于卤代甲基三嗪的光引发剂的实例是2
‑
[2
‑
(4
‑
甲氧基
‑
苯基)
‑
乙烯基]
‑
4,6
‑
双
‑
三氯甲基[1,3,5]三嗪,2
‑
(4
‑
甲氧基
‑
苯基)
‑
4,6
‑
双
‑
三氯甲基[1,3,5]三嗪,2
‑
(3,4
‑
二甲氧基苯基)
‑
4,6
‑
双
‑
三氯甲基[1,3,5]三嗪,2
‑
甲基
‑
4,6
‑
双
‑
三氯甲基[1,3,5]三嗪。
[0138]
当本发明的可光固化的组合物用于混合体系(在这方面是指自由基和阳离子固化体系的混合物)时,阳离子光引发剂也可用作其他光引发剂e)。合适的阳离子光引发剂的实例是在美国专利号4,950,581描述的芳族锍盐、鏻盐或錪盐,或者环戊二烯基芳烃
‑
铁(ii)络盐,例如在gb2348644,美国专利号4,450,598和4,136,055,wo 00/10972以及wo 00/26219中描述的(η6‑
异丙基苯)(η5‑
环戊二烯基)六氟磷酸铁(ii)或基于肟的光潜酸。
[0139]
添加剂f)可以是例如加速剂/共引发剂、热引发剂、粘合剂、稳定剂及其混合物。
[0140]
本发明的可光固化的组合物还可以方便地包括加速剂/共引发剂,例如醇、硫醇、硫醚、胺或醚,其具有与邻近杂原子的碳键合的可用氢,还包括二硫化物和膦,如ep 438123和gb 2180358中描述的那些。这种加速剂/共引发剂的量通常在0.2至15重量%之间,优选为0.2至8重量%之间。
[0141]
胺加速剂/共引发剂的合适实例包括但不限于:脂族、脂环族、芳族、芳基
‑
脂族、杂环、低聚或聚合胺。它们可以是伯胺、仲胺或叔胺,例如丁胺、二丁胺、三丁胺、环己胺、芐基二甲胺、二环己胺、n
‑
苯基甘氨酸、三乙基胺、苯基
‑
二乙醇胺、三乙醇胺、哌啶、哌嗪、吗啉、吡啶、喹啉、二甲基氨基苯甲酸的酯、米氏酮(4,4'
‑
双
‑
二甲基氨基二苯甲酮)和相应的衍生物。
[0142]
作为胺促进剂/共引发剂,可用使用胺改性的丙烯酸酯化合物:这些胺改性的丙烯酸酯化合物的实例包括与伯胺或仲胺反应而被改性的丙烯酸酯,如us 3,844,916、ep 280222、us 5,482,649或us 5,734,002中描述的。
[0143]
多官能胺和聚合物胺衍生物也适合作为共引发剂,一些实例为来自igm resins b.v.的asa、来自rahn a.g.的ab
‑
2以及来自lambson limited的7040,或者us2013/0012611中描述的那些。
[0144]
根据本发明的固化过程,特别是在颜料组合物的情况下,也可以通过加入热引发剂作为额外添加剂f)进行协助,该组分是在热条件下形成自由基的化合物,例如偶氮化合物(诸如2,2'
‑
偶氮二(4
‑
甲氧基
‑
2,4
‑
二甲基戊腈))、三氮烯、二偶氮硫化物、五氮二烯或过氧化合物(例如在ep 245639中描述的过氧化氢或过碳酸酯,如叔丁基过氧化氢)。
[0145]
也可以将粘结剂加入到本发明的可光固化的组合物中。当可光固化化合物是液体或粘性物质时加入粘结剂是特别有利的。粘结剂的量可以是例如5至60重量%,优选为10至50重量%。根据使用领域和对于其要求的性能,如在含水和有机溶剂体系中的可展性(developability)、对基材的粘合力和对氧的敏感性,进行粘结剂的选择。
[0146]
合适的粘结剂是例如分子量大约为5000至2 000 000da,优选为10 000至1 000000da的聚合物。示意性实例是:丙烯酸酯和甲基丙烯酸酯的均聚物和共聚物,例如甲基丙烯酸甲酯/丙烯酸乙酯/甲基丙烯酸的共聚物、聚(甲基丙烯酸烷基酯)、聚(丙烯酸烷基酯);纤维素酯和醚,诸如乙酸纤维素、乙酸丁酸纤维素、甲基纤维素、乙基纤维素;聚乙烯醇缩丁醛、聚乙烯醇缩甲醛、环化橡胶,聚醚如聚环氧乙烷、聚环氧丙烷、聚四氢呋喃;聚苯乙烯、聚碳酸酯、聚氨酯;氯化聚烯烃,例如聚氯乙烯、氯乙烯/偏二氯乙烯的共聚物、偏二氯乙烯与丙烯腈、甲基丙烯酸甲酯和乙酸乙烯酯的共聚物、聚乙酸乙烯酯、共聚(乙烯/乙酸乙烯酯)、诸如聚己内酰胺和聚(己二酰己二胺)的聚合物、诸如聚(对苯二甲酸乙二醇酯)和聚(六亚甲基二醇琥珀酸酯)的聚酯。
[0147]
合适的稳定剂是例如热抑制剂,诸如氢醌、氢醌衍生物、对甲氧基苯酚、β
‑
萘酚或位阻酚,如2,6
‑
二(叔丁基)
‑
对甲酚,其可防止过早聚合。为增加暗贮存稳定性,可以使用例如铜化合物(诸如环烷酸铜、硬脂酸铜或辛酸铜)、磷化合物(例如三苯基膦、三丁基膦、亚磷酸三乙酯、亚磷酸三苯酯或亚磷酸三苄酯)、季铵化合物(例如四甲基氯化铵或三甲基苄基氯化铵)、或羟基胺衍生物(例如n,n
‑
二乙基羟基胺)。出于在聚合期间隔绝大气氧的目的,可以加入石蜡或类似的蜡状物质,它们不溶于聚合物,在聚合开始时迁移到表面并形成透明表面层,该表面层防止空气进入。
[0148]
还可能的是加入光稳定剂,诸如uv吸收剂,例如羟苯基苯并三唑、羟苯基二苯甲酮、草酸酰胺或羟苯基
‑
s
‑
三嗪型的那些。这些化合物可以按它们自身使用或以混合物的形式使用,使用或不使用位阻胺(hals)。
[0149]
根据本发明的可光固化的组合物也可包括作为另外的添加剂f)的可光还原染料,例如呫吨、苯并呫吨、苯并硫代呫吨、噻嗪、焦宁、卟啉或吖啶染料,和/或可辐射断裂的三卤代甲基化合物。这些化合物描述于例如ep445624中。
[0150]
取决于所需的用途,另外的常用添加剂f)是荧光增白剂、填料、颜料(白色和着色颜料两者)、着色剂、抗静电剂、润湿剂或流动改进剂。也可以使用本领域常用的添加剂,例如抗静电剂、流动改进剂和粘附增强剂。
[0151]
也可以向本发明的组合物中加入本领域常用的链转移试剂。实例包括硫醇、胺和苯并三唑。
[0152]
本发明的组合物也可包含着色剂和/或着色颜料。取决于所需的用途,可以使用无机和有机颜料两者。这样的添加剂是本领域技术人员已知的;一些实例是炭黑、氧化铁,如氧化铁黄、氧化铁红、铬黄、铬绿、镍钛黄、群青、钴蓝、钒酸铋、镉黄和镉红。有机颜料的实例是单或双偶氮颜料及其金属配合物、酞菁颜料、多环颜料,如苝、蒽醌、硫代靛蓝、喹吖啶酮
或三苯基甲烷颜料、以及二酮
‑
吡咯并
‑
吡咯、异二氢吲哚酮,如四氯异二氢吲哚酮、异二氢吲哚、二噁嗪、苯并咪唑啉酮和喹啉并酞酮颜料。颜料可以按自身或以混合物用于制剂。
[0153]
取决于所需的用途,可以向制剂中以本领域常用的量加入颜料,例如以基于总质量0.1至30重量%或10至25重量%的量。
[0154]
组合物也可包括例如特别宽种类的有机着色剂。实例是偶氮染料、次亚甲基染料、蒽醌染料和金属配合物染料。通常的浓度是例如基于总质量的0.1至20重量%,特别地1至5重量%。
[0155]
添加剂的选择由所相关的应用领域和该领域需要的性能支配。以上所述的添加剂f)是本领域已知的,且因此以本领域通常的量使用。
[0156]
本发明的可光固化的组合物适合用于各种目的,例如作为印刷油墨,如丝网印刷油墨、苯胺印刷油墨或胶版印刷油墨,作为透明涂层,作为着色涂层,例如用于木材或金属,作为粉末涂料,作为尤其用于纸、木材、金属或塑料的油漆材料,作为可日光固化油漆用于标记结构物和道路、用于光刻再现工艺、用于全息摄影记录材料、用于图像记录工艺或用于印刷板的生产,该印刷板可以使用有机溶剂或使用含水碱性介质显影、用于丝网印刷用掩模的生产,作为牙科填充化合物,作为粘合剂,作为压敏粘合剂,作为层压树脂,作为光刻胶,例如电抗蚀剂、蚀刻抗蚀剂或永久抗蚀剂,两者均为液体且为干燥膜的形式,作为可光构造电介质,和作为电子电路的焊剂掩模、作为抗蚀剂,用于任何类型显示屏用滤色器制造中或用于等离子体显示器和电致发光显示器制造期间结构的形成、用于光学开关、光栅(干扰光栅)的制造、用于由本体固化(在透明模具中的uv固化)或根据立体平版印刷方法的三维制品的制造(例如描述于us 4,575,330)、用于复合材料(例如可包括玻璃纤维和/或其它纤维和其它助剂的苯乙烯聚酯)的制造、以及用于本领域技术人员熟知的三维印刷的其他方法、用于电子组件的涂敷或密封或作为光纤的涂料。
[0157]
本发明的可光固化的组合物也适用于光学透镜例如隐形眼镜或菲涅耳透镜的制造,用于医疗设备、辅助器或植入物的制造,以及用于干燥膜油漆的制造。
[0158]
本发明的可光固化的组合物还适用于具有热致性能的凝胶的制备。这样的凝胶描述于例如de 19700064和ep 678 534。
[0159]
根据本发明的化合物和组合物也可以用作自由基光引发剂或可辐射固化粉末涂料的光引发体系。
[0160]
本发明的可光固化的组合物适合于用作例如所有种类基材的涂料材料,基材例如木材、纺织品、纸、陶瓷、玻璃、塑料诸如聚酯、聚对苯二甲酸乙二醇酯、聚烯烃或乙酸纤维素,特别是以膜的形式、以及金属诸如al、cu、ni、fe、zn、mg或co和gaas、si或sio2,该基材待涂布保护层或例如通过成像曝光施加图像。
[0161]
包含至少一种式(ia)和/或(ii)化合物的可光固化的组合物代表本发明的另一个主题。
[0162]
根据另一方面,本发明的再一个主题是一种使可光聚合的组合物和油墨光固化的方法,所述方法包括以下步骤:
[0163]
i)制备可光聚合的组合物,所述组合物包含:
[0164]
‑
如上文所定义的组分a)、b)和c);或者
[0165]
‑
如上文所定义的组分a)和d);
[0166]
ii)将所述可光聚合的组合物涂覆或印刷在基材上,以及
[0167]
iii)用光源使在所述基材上涂覆或印刷的所述组合物光聚合。
[0168]
因此,可以使用大量各种各样的光源,光源的发射波长为约200nm至约600nm。点光源和平面状投射器(灯毯)都是合适的。实例是:碳弧灯,氙弧灯,中压、高压和低压汞弧辐射体,适当时掺杂有金属卤化物(金属卤化物灯),微波激发金属蒸气灯,受激准分子灯,超光化荧光管,荧光灯,氩白炽灯,电子闪光灯,摄影照明灯,发光二极管(led),电子束、x射线和激光。在灯和要曝光的根据本发明的基材之间的距离可根据预期用途和灯的类型和强度而变化,并且可以是例如1cm至150cm。
[0169]
特别优选的是发射波长在365nm至420nm之间、优选为365nm、385nm和395nm的led光源。
[0170]
所述可光聚合的组合物还可被施加到已经包含了涂层或印刷层的基材上。在用所述光源进行光聚合之后,在所述可光聚合的组合物上可被套印或涂布一种或多种适于印刷或涂覆的组合物。
[0171]
包含至少一种式(ia)和/或(ii)化合物的任何制品代表本发明的另一主题。
[0172]
本发明的另一个主题是通过如下方法获得的制品:通过所述涂覆或印刷装置将所述可光聚合的组合物施加在所述基材上,并通过所述光源进行光聚合,对所述制品不作进一步精加工,或再进行涂覆或印刷对所述制品进一步精加工。
[0173]
令人惊讶地,我们发现式(ia)和(ii)所示的化合物与macromolecules 2009,42,6031
‑
6037(pi
‑
1)描述的化合物相比,在透明和着色系统的光聚合过程中展现更高的反应性。此外,我们首次证明了能够合成式(i)和(ii)化合物,这些化合物的光谱活性得到提高。
[0174]
例如,我们发现在以相同的量使用时,实施例5描述的式(ia)的化合物展现反应性是macromolecules 2009,42,6031
‑
6037(pi
‑
1)描述的参考化合物的2倍,此外,异丙基噻吨酮以这种方式在led波长(例如400nm)下工作能够合成该化合物。
[0175]
在以下实施例中,仅用于说明而非限制性目的,记载了本发明式(ia)和(ii)的α
‑
二甲硅烷氧基酮和可光固化的组合物的示意性制备。
[0176]
实施例
[0177]
定义和仪器
[0178]1h nmr图谱是使用bruker avance 400mhz或bruker dmx 500mhz或bruker dmx 600mhz记录的。
[0179]
红外图谱是使用jasco的ft
‑
ir 430记录的。
[0180]
下列实施例中,化学结构与标示的化学名称不一致时,以化学结构为准。
[0181]
实施例1
[0182]1‑
(4
‑
甲氧基苯基)
‑2‑
甲基
‑2‑
((1,1,2,2,2
‑
五甲基二硅烷基)氧基)丙
‑1‑
酮的合成
[0183][0184]
保持温度在0℃至5℃下,在约30分钟内向5g(46.2mmol)苯甲醚和11.16g(48.5mmol)α
‑
溴代异丁酰溴在70ml二氯甲烷中的溶液加入6.46g(48.5mmol)三氯化铝。在
室温下2小时后,将反应混合物倒入100ml冰水和2ml 37%hcl的混合物中。分离有机相并用水洗涤,用硫酸钠干燥,过滤。将该溶液用120ml 0.5m氢氧化钾在甲醇中的溶液回流处理3小时。用100ml水和50ml二氯甲烷稀释后,分离两相。有机相用水洗涤,硫酸钠干燥,过滤,真空蒸发溶剂。粗产物无需任何纯化即可用于下一步骤。
[0185]
将2
‑
羟基
‑1‑
(4
‑
甲三甲)
‑2‑
甲基丙
‑1‑
酮(2g,10.2mmol)和氯代五甲基二硅烷(1.7g,10.2mmol)溶于10ml二氯甲烷中,加入0.69g(10.2mmol)咪唑。将反应混合物在室温搅拌3小时,然后真空蒸发。粗产物在20ml甲苯中稀释并经硅胶过滤。真空除去溶剂,得到纯产物(1.9g)。
[0186]1h nmr(cdcl3,δppm):0.00(s,9h),0.13(s,6h),1.55(s,6h),3.85(s,3h),6.88(d,2h),8.22(d,2h)。
[0187]
实施例2
[0188]2‑
甲基
‑1‑
(4
‑
(甲基硫代)苯基)
‑2‑
((1,1,2,2,2
‑
五甲基二硅烷基)氧基)丙
‑1‑
酮的合成
[0189][0190]
保持温度在0℃至5℃下,在约30分钟内向5g(40.2mmol)硫苯甲醚和9.72g(42.2mmol)α
‑
溴代异丁酰溴在70ml二氯甲烷中的溶液加入5.62g(42.2mmol)三氯化铝。在室温下2小时后,将反应混合物倒入100ml冰水和2ml 37%hcl的混合物中。分离有机相并用水洗涤,用硫酸钠干燥,过滤。将该溶液用100ml 0.5m氢氧化钾在甲醇中的溶液回流处理3小时。用100ml水和50ml二氯甲烷稀释后,分离两相。有机相用水洗涤,硫酸钠干燥,过滤,真空蒸发溶剂。粗产物无需任何纯化即可用于下一步骤。
[0191]
将2
‑
羟基
‑2‑
甲基
‑1‑
(4
‑
(甲基硫代)苯基)丙
‑1‑
酮(1.4g,6.6mmol)和氯代五甲基二硅烷(1.1g,6.6mmol)溶于10ml二氯甲烷中,加入0.44g(6.6mmol)咪唑。将反应混合物在室温搅拌3小时,然后真空蒸发。粗产物在20ml甲苯中稀释并经硅胶过滤。真空除去溶剂,得到纯产物(1.35g)。
[0192]1h nmr(cdcl3,δppm):0.00(s,9h),0.14(s,6h),1.53(s,6h),2.50(s,3h),7.2(d,2h),8.14(d,2h)。
[0193]
实施例3
[0194]2‑
甲基
‑1‑
(4
‑
吗啉代苯基)
‑2‑
((1,1,2,2,2
‑
五甲基二硅烷基)氧基)丙
‑1‑
酮的合成
[0195][0196]
保持温度在0℃至5℃下,在约30分钟内向10g(104mmol)氟苯和25.1g(109mmol)α
‑
溴代异丁酰溴在120ml二氯甲烷中的溶液加入14.5g(109mmol)三氯化铝。在室温下2小时后,将反应混合物倒入100ml冰水和2ml 37%hcl的混合物中。分离有机相并用水洗涤,用硫
酸钠干燥,过滤。将该溶液用100ml 0.5m氢氧化钾在甲醇中的溶液回流处理3小时。用100ml水和50ml二氯甲烷稀释后,分离两相。有机相用水洗涤,硫酸钠干燥,过滤,真空蒸发溶剂。将粗产物(4g,21.3mmol)、吗啉(1.85g,21.3mmol)和碳酸钾(4.42g,32mmol)溶解在50ml二甲亚砜中。保持在160℃18小时后,将悬浮液冷却,用水稀释并用二氯甲烷萃取。有机相用水洗涤,硫酸钠干燥,过滤,真空蒸发溶剂。粗产物无需任何纯化即可用于下一步骤。
[0197]
将2
‑
羟基
‑2‑
甲基
‑1‑
(4
‑
吗啉代苯基)丙
‑1‑
酮(1.67g,6.69mmol)和氯代五甲基二硅烷(1.11g,6.69mmol)溶于30ml二氯甲烷中,加入0.45g(6.69mmol)咪唑。将反应混合物在室温搅拌3小时,然后真空蒸发。粗产物经快速柱色谱法(二氯甲烷/乙酸乙酯9:1)纯化,获得1.15g纯产物。
[0198]1h nmr(cdcl3,δppm):0.01(s,9h),0.14(s,6h),1.50(s,6h),3.29(t,2h),3.86(t,2h),6.83(d,2h),8.19(d,2h)。
[0199]
实施例4
[0200]2‑
甲基
‑2‑
((1,1,2,2,2
‑
五甲基二硅烷基)氧基)
‑1‑
(4
‑
苯氧基苯基)丙
‑1‑
酮的合成
[0201][0202]
保持温度在0℃至5℃下,在约30分钟内向15g(88.1mmol)二苯醚和10.11g(44mmol)α
‑
溴代异丁酰溴在100ml二氯甲烷中的溶液加入5.87g(44mmol)三氯化铝。在室温下2小时后,将反应混合物倒入100ml冰水和2ml 37%hcl的混合物中。分离有机相并用水洗涤,用硫酸钠干燥,过滤。将该溶液用120ml 0.5m氢氧化钾在甲醇中的溶液回流处理3小时。用100ml水和50ml二氯甲烷稀释后,分离两相。有机相用水洗涤,硫酸钠干燥,过滤,真空蒸发溶剂。粗产物经快速柱色谱法(二氯甲烷)纯化,用于下一步骤。
[0203]
将2
‑
羟基
‑2‑
甲基
‑1‑
(4
‑
苯氧基苯基)丙
‑1‑
酮(1.5g,5.8mmol)和氯代五甲基二硅烷(0.97g,5.8mmol)溶于20ml二氯甲烷中,加入0.4g(5.8mmol)咪唑。将反应混合物在室温搅拌3小时,然后真空蒸发。粗产物用20ml甲苯稀释并经硅胶过滤。真空除去溶剂,得到纯产物(1.27g)。
[0204]1h nmr(cdcl3,δppm):0.05(s,9h),0.15(s,6h),1.55(s,6h),6.95(d,2h),7.07(d,2h),7.18(t,1h),7.38(t,2h),8.20(d,2h)。
[0205]
实施例5
[0206]2‑
甲基
‑2‑
((1,1,2,2,2
‑
五甲基二硅烷基)氧基)
‑1‑
(4
‑
(苯基硫代)苯基)丙
‑1‑
酮的合成
[0207][0208]
保持温度在0℃至5℃下,在约30分钟内向15g(80.5mmol)二苯基硫醚和9.25g(40.2mmol)α
‑
溴代异丁酰溴在100ml二氯甲烷中的溶液加入5.35g(40.2mmol)三氯化铝。在室温下2小时后,将反应混合物倒入100ml冰水和2ml 37%hcl的混合物中。分离有机相并用
水洗涤,用硫酸钠干燥,过滤。将该溶液用100ml 0.5m氢氧化钾在甲醇中的溶液回流处理3小时。用100ml水和50ml二氯甲烷稀释后,分离两相。有机相用水洗涤,硫酸钠干燥,过滤,真空蒸发溶剂。粗产物经快速柱色谱法(二氯甲烷)纯化,用于下一步骤。
[0209]
将2
‑
羟基
‑2‑
甲基
‑1‑
(4
‑
(苯基硫代)苯基)丙
‑1‑
酮(1.5g,5.5mmol)和氯代五甲基二硅烷(0.92g,5.5mmol)溶于20ml二氯甲烷中,加入0.37g(5.5mmol)咪唑。将反应混合物在室温搅拌3小时,然后真空蒸发。粗产物用20ml甲苯稀释并经硅胶过滤。真空除去溶剂,得到纯产物(1.30g)。
[0210]1h nmr(cdcl3,δppm):0.00(m,9h),0.14(m,6h),1.54(s,6h),7.18(d,2h),7.37(m,3h),7.47(d,2h),8.08(d,2h)。
[0211]
实施例6
[0212]2‑
甲基
‑2‑
((1,1,2,2,2
‑
五甲基二硅烷基)氧基)
‑
3,4
‑
二氢萘
‑
1(2h)
‑
酮的合成
[0213][0214]
步骤1.在氮气气氛下,边搅拌边将1
‑
四氢萘酮(tetralone)(4.55ml,34.202mmol)在20ml thf中的溶液滴加入nah(60%在油中的分散液,2.73g,68.404mmol)和碳酸二甲酯(5.76ml,68.404mmol)在thf(80ml)中的悬液中。反应加热回流2小时。然后冷却至室温,用1m盐酸水溶液酸化,用乙酸乙酯(2x50ml)萃取。有机层用10%nahco3水溶液、盐水洗涤,硫酸钠干燥,减压除去溶剂。将残留物溶解在dmf(20ml)中,边搅拌边滴加入nah(60%在油中的分散液,2.05g,51.3mmol)在dmf(50ml)中的悬液中。在氮气气氛下将混合物在室温下搅拌1小时。然后加入碘甲烷(3.19ml,51.3mmol),混合物在同一温度下再搅拌1小时。然后加入水,混合物用乙酸乙酯(2x50ml)萃取。有机层用盐水洗涤,经硫酸钠干燥并在减压下除去溶剂。将残留物溶解在乙酸(50ml)中并加入37%hcl(11ml)。将混合物加热回流4小时,冷却至室温,用水(100ml)稀释,用二氯甲烷(2x50ml)萃取。有机层用盐水洗涤,硫酸钠干燥,减压除去溶剂。通过快速柱色谱法(乙酸乙酯/石油醚=5/95)纯化粗产物,获得5.056g纯产物。
[0215]1h nmr(cdcl3,δppm):1.24
–
1.30(d,3h),1.84
–
1.92(m,1h),2.16
–
2.23(m,1h),2.55
–
2.62(m,1h),2.94
–
3.07(m,2h),7.21(d,1h),7.27
–
7.32(t,1h),7.43
–
7.48(m,1h),8.01
–
8.06(m,1h)。
[0216]
步骤2.向nai(3.567g,23.84mmol)在乙腈(50ml)中的悬液加入2
‑
甲基
‑
3,4
‑
二氢萘
‑
1(2h)
‑
酮(2.728g,17.03mmol,步骤1制备)在乙腈(10ml)中的溶液,再加入三乙胺(3.55ml 25.54mmol)。然后将混合物冷却至0℃,并滴加入氯三甲基硅烷(2.81ml,22.14mmol)。混合物在室温和氮气气氛下搅拌2小时。然后加入水(50ml),混合物用乙酸乙酯(2
×
25ml)萃取。有机层用盐水洗涤,经硫酸钠干燥,并减压除去溶剂。将残留物溶解于二氯甲烷(20ml),在0℃下加入3
‑
氯过苯甲酸(3.82g,22.13mmol)。混合物在室温下搅拌30分钟。然后,用10%nahco3水溶液、盐水洗涤,用硫酸钠干燥并在减压下除去溶剂。将残留物溶解在thf(40ml),加入四
‑
正丁基氟化铵(6.68g,25.54mmol)。将反应物在室温下搅拌过夜。然后加入水(50ml),混合物用乙酸乙酯(2x50ml)萃取。有机层用10%nahco3水溶液、盐水洗涤,用硫酸钠干燥并在减压下除去溶剂。将粗产物通过快速柱色谱法纯化(乙酸乙酯/石油
醚=1/5)纯化,获得2.177g纯产物。
[0217]1h nmr(cdcl3,δppm):1.4(s,3h),2.17
–
2.28(m,2h),2.99
–
3.14(m,2h),7.23
–
7.28(m,1h),7.32
–
7.37(t,1h),7.49
–
7.55(m,1h),8.01
–
8.06(m,1h)。
[0218]
步骤3.向2
‑
羟基
‑2‑
甲基
‑
3,4
‑
二氢萘
‑
1(2h)
‑
酮(300mg,1.704mmol,步骤2制备)在二氯甲烷(10ml)中的溶液加入氯代五甲基二硅烷(494μl,2.56mmol)和咪唑(174mg,2.56mmol)。混合物在室温下搅拌3小时,然后减压浓缩。残留物通过硅胶短垫过滤(环己烷100%2cv,然后用乙酸乙酯/环己烷5/95洗脱),得到90mg纯产物。
[0219]1h nmr(500mhz,cdcl3):0.09(s,9h),0.18(s,3h),0.20(s,3h),1.41(s,3h),2.01
–
2.08(m,1h),2.19
–
2.26(m,1h),2.84
–
2.91(m,1h),3.10
–
3.17(m,1h),7.18
–
7.22(d,1h),7.27
–
7.32(t,1h),7.43
–
7.47(m,1h),7.99
–
8.02(m,1h)。
[0220]
实施例7
[0221]6‑
甲氧基
‑2‑
甲基
‑2‑
[(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基]
‑
1,2,3,4
‑
四氢萘
‑1‑
酮的合成
[0222][0223]
步骤1.在氮气气氛下,边搅拌边将6
‑
甲氧基
‑1‑
四氢萘酮(5g,28.37mmol)在50ml thf中的溶液滴加入nah(60%在油中的分散液,2.27g,56.75mmol)和碳酸二甲酯(4.78ml,56.75mmol)在thf(50ml)中的悬液中。反应加热回流2小时。然后冷却至室温,用1m盐酸水溶液酸化,用乙酸乙酯(2x50ml)萃取。有机层用10%nahco3水溶液、盐水洗涤,硫酸钠干燥,减压除去溶剂。将残留物溶解在dmf(20ml)中,边搅拌边滴加入nah(60%在油中的分散液,1.7g,42.55mmol)在dmf(50ml)中的悬液中。在氮气气氛下将混合物在室温下搅拌1小时。然后加入碘甲烷(2.6ml,42.55mmol),混合物在同一温度下再搅拌1小时。然后加入水,混合物用乙酸乙酯(2x50ml)萃取。有机层用盐水洗涤,经硫酸钠干燥并在减压下除去溶剂。将残留物溶解在乙酸(50ml)中并加入37%hcl(11ml)。将混合物加热回流4小时,冷却至室温,用水(100ml)稀释,用二氯甲烷(2x50ml)萃取。有机层用盐水洗涤,硫酸钠干燥,减压除去溶剂。通过快速柱色谱法(乙酸乙酯/石油醚=1/9)纯化粗产物,获得5.04g纯产物(产率=93%)。
[0224]1h nmr(500mhz,cdcl3):δ=1.22
–
1.27(d,3h),1.80
–
1.89(m,1h),2.13
–
2.19(m,1h),2.49
–
2.56(m,1h),2.88
–
3.02(m,2h),3.83(s,3h),6.65
–
6.67(m,1h),6.79
–
6.82(m,1h),7.98
–
8.01(m,1h)。
[0225]
步骤2.向nai(2.62g,17.47mmol)在乙腈(25ml)中的悬液加入6
‑
甲氧基
‑2‑
甲基
‑
1,2,3,4
‑
四氢萘
‑1‑
酮(步骤1制备)(2g,12.48mmol)在乙腈(10ml)中的溶液,再加入三乙胺(2.6ml 18.72mmol)。然后将混合物冷却至0℃,并滴加入氯三甲基硅烷(2.06ml,16.22mmol)。混合物在室温和氮气气氛下搅拌2小时。然后加入水(25ml),混合物用乙酸乙酯(2x25ml)萃取。有机层用盐水洗涤,经硫酸钠干燥,并减压除去溶剂。将残留物溶解于二氯甲烷(10ml),在0℃下加入3
‑
氯过苯甲酸(3.635g,21.06mmol)。混合物在室温下搅拌30分钟。然后用10%nahco3水溶液、盐水洗涤,用硫酸钠干燥并在减压下除去溶剂。将残留物溶解在thf(40ml),加入四
‑
正丁基氟化铵(3.94g,12.48mmol)。将反应物在室温下搅拌过夜。
然后加入水(50ml),混合物用乙酸乙酯(2x50ml)萃取。有机层用10%nahco3水溶液、盐水洗涤,用硫酸钠干燥并在减压下除去溶剂。将粗产物通过快速柱色谱法纯化(乙酸乙酯/石油醚=3/7)纯化,获得557mg纯产物(产率=22%)。
[0226]1h nmr(500mhz,cdcl3):δ=1.4(s,3h),2.15
–
2.26(m,2h),2.92
–
3.1(m,2h),3.85(s,3h),6.72(s,1h),6.84
–
6.88(m,1h),7.97
–
8.01(m,1h)。
[0227]
步骤3.向2
‑
羟基
‑6‑
甲氧基
‑2‑
甲基
‑
1,2,3,4
‑
四氢萘
‑1‑
酮(步骤2制备)(270g,1.31mmol)在二氯甲烷(2ml)中的溶液加入氯代五甲基二硅烷(303μl,1.57mmol)和咪唑(107mg,1.57mmol)。混合物在室温下搅拌3小时,然后减压浓缩。残留物通过氧化铝短垫过滤(石油醚2cv,然后乙酸乙酯/石油醚5/95),得到150mg纯产物(产率=34%)。
[0228]1h nmr(300mhz,dmso
‑
d6):
‑
0.01(s,9h),0.15(s,3h),0.13(s,3h),1.32(s,3h),1.96
–
2.15(m,2h),2.82
–
2.96(m,1h),2.97
–
3.10(m,1h),3.83(s,3h),6.83
–
6.87(d,1h),6.88
–
6.95(dd,1h),7.81
–
7.88(d,1h)。
[0229]
实施例8
[0230]2‑
甲基
‑1‑
[4
‑
(4
‑
{2
‑
甲基
‑2‑
[(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基]丙酰基}苯氧基)苯基]
‑2‑
[(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基]丙
‑1‑
酮(式(i)/(ia),n=2;ar(ar’)=
‑
亚苯基
‑
o
‑
亚苯基
‑
)的合成
[0231][0232]
将2
‑
羟基
‑1‑
{4
‑
[4
‑
(2
‑
羟基
‑2‑
甲基丙酰基)苯氧基]苯基}
‑2‑
甲基丙
‑1‑
酮(2g,5.841mmol)溶解在二氯甲烷(10ml)中,加入氯五甲基二硅烷(2.71ml,14.02mmol)和咪唑(954mg,14.02mmol)。混合物在室温下搅拌2小时,然后减压浓缩。粗产物通过快速色谱法(石油醚2cv;乙酸乙酯/石油醚5/95)纯化,得到2.4g纯产物(产率=69%)。
[0233]1h nmr(300mhz,dmso
‑
d6):
‑
0.03(s,18h),0.15(s,12h),1.5(s,12h),7.10
–
7.18(d,4h),8.13
–
8.20(d,4h)。
[0234]
实施例9
[0235]1‑
[(1
‑
苯甲酰基环己基)氧基]
‑
1,1,2,2,2
‑
五甲基二硅烷的合成
[0236][0237]
将1
‑
苯甲酰基环己
‑1‑
醇(1g,4.89mmol)溶解在二氯甲烷(10ml)中,加入氯五甲基
二硅烷(1.14ml,5.87mmol)和咪唑(400mg,5.87mmol)。混合物在室温下搅拌2小时,然后减压浓缩。粗产物通过快速氧化铝色谱法(石油醚2cv;乙酸乙酯/石油醚3/97)纯化,得到350mg纯产物(产率=21%)。
[0238]1h nmr(300mhz,dmso
‑
d6):0.00(s,9h),0.08(s,6h),1.21
‑
1.54(m,4h),1.55
‑
1.71(m,2h),1.71
‑
1.83(m,2h),1.86
‑
2.00(m,2h),7.44
‑
7.54(m,2h),7.54
‑
7.64(m,1h),8.02
‑
8.11(m,2h)。
[0239]
实施例10
[0240]6‑
甲基
‑6‑
[(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基]
‑
6,7,8,9
‑
四氢
‑
5h
‑
苯并[7]轮烯
‑5‑
酮的合成
[0241][0242]
步骤1.在氮气气氛下边搅拌边将1
‑
苯并环庚酮(5g,31.21mmol)在50ml thf中的溶液滴加入nah(60%在油中的分散液,2.50g,62.42mmol)和碳酸二甲酯(5.26ml,62.42mmol)在thf(50ml)中的悬液中。将反应加热回流2小时。然后冷却至室温,用1m盐酸水溶液酸化,用乙酸乙酯(2x50ml)萃取。有机层用10%nahco3水溶液、盐水洗涤,硫酸钠干燥,减压除去溶剂。将残留物溶解在dmf(20ml)中,边搅拌边将该溶液滴加入nah(60%在油中的分散液,1.87g,46.8mmol)在dmf(50ml)中的悬液中。混合物在室温和氮气气氛下搅拌1小时。然后加入碘甲烷(2.91ml,46.8mmol),并将混合物在同一温度下再搅拌1小时。然后加入水,混合物用乙酸乙酯(2x50ml)萃取。有机层用盐水洗涤,经硫酸钠干燥,减压下除去溶剂。将残留物溶解在乙酸(50ml)中,加入37%hcl(11ml)。混合物回流加热4小时,冷却至室温,用水(100ml)稀释,并用二氯甲烷(2x50ml)萃取。有机层用盐水洗涤,硫酸钠干燥,减压除去溶剂。粗产物经快速色谱纯化(乙酸乙酯/石油醚=5/95),得到4.97g纯产物(产率=91%)。
[0243]1h nmr(500mhz,cdcl3):δ=1.19
–
1.24(d,3h),1.54
–
1.64(m,1h),1.66
–
1.74(m,1h),1.86
–
1.95(m,1h),2.02
–
2.12(m,1h),2.87
–
3.04(m,3h),7.18
–
7.21(d,1h),7.25
–
7.29(d,1h),7.34
–
7.39(m,1h),7.64
–
7.68(m,1h)。
[0244]
步骤2.向nai(5.98g,39.93mmol)在乙腈(50ml)中的悬液加入6
‑
甲基
‑
6,7,8,9
‑
四氢
‑
5h
‑
苯并[7]轮烯
‑5‑
酮(步骤1制备)(4.97g,28.52mmol)在乙腈(10ml)中的溶液,再加入三乙胺(5.95ml 42.78mmol)。然后将混合物冷却至0℃,并滴加入氯三甲基硅烷4.71ml,37.08mmol)。混合物在室温和氮气气氛下搅拌2小时。然后加入水(50ml),混合物用乙酸乙酯(2x25ml)萃取。有机层用盐水洗涤,经硫酸钠干燥,并减压除去溶剂。将残留物溶解于二氯甲烷(20ml),在0℃下加入3
‑
氯过苯甲酸(8.31g,48.15mmol)。混合物在室温下搅拌30分钟。然后用10%nahco3水溶液、盐水洗涤,用硫酸钠干燥并在减压下除去溶剂。将残留物溶解在thf(40ml),加入四
‑
正丁基氟化铵(7.46g,28.52mmol)。将反应物在室温下搅拌过夜。然后加入水(50ml),混合物用乙酸乙酯(2x50ml)萃取。有机层用10%nahco3水溶液、盐水洗涤,用硫酸钠干燥并在减压下除去溶剂。将粗产物通过快速色谱法纯化(乙酸乙酯/石油醚=1/9)纯化,获得3.654g纯产物(产率=67%)。
[0245]1h nmr(500mhz,cdcl3):δ=1.25(s,3h),1.88
–
2.05(m,3h),2.11
–
2.18(m,1h),
2.83
–
2.88(m,2h),7.14
–
7.17(m,1h),7.24
–
7.28(m,1h),7.35
–
7.39(m,2h)。
[0246]
步骤3.向6
‑
羟基
‑6‑
甲基
‑
6,7,8,9
‑
四氢
‑
5h
‑
苯并[7]轮烯
‑5‑
酮(步骤2制备)(1g,5.26mmol)在二氯甲烷(50ml)中的溶液加入氯五甲基二硅烷(1.22ml,6.31mmol)和咪唑(429mg,6.31mmol)。混合物在室温下搅拌3小时,然后减压浓缩。残留物通过氧化铝短垫过滤(石油醚4cv),得到506mg纯产物(产率=30%)。
[0247]1h nmr(300mhz,dmso
‑
d6):
‑
0.10(s,9h),0.05(s,3h),0.16(s,3h),1.36(s,3h),1.59
–
1.75(m,1h),1.76
–
1.87(m,1h),1.87
–
2.06(m,2h),2.69
–
2.81(m,1h),2.86
–
2.99(m,1h),7.17
–
7.24(m,1h),7.24
–
7.30(m,2h),7.32
–
7.41(m,1h)。
[0248]
实施例11
[0249]
主要基于如下化合物的混合物:2,3
‑
二氢
‑6‑
[2
‑
(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基
‑2‑
甲基
‑1‑
氧代丙基]
‑
1,1,3
‑
三甲基
‑3‑
[4
‑
(2
‑
(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基
‑2‑
甲基
‑1‑
氧代丙基)苯基]
‑
1h
‑
茚;2,3
‑
二氢
‑5‑
[2
‑
(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基
‑2‑
甲基
‑1‑
氧代丙基]
‑
1,1,3
‑
三甲基
‑3‑
[4
‑
(2
‑
(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基
‑2‑
甲基
‑1‑
氧代丙基)苯基]
‑
1h
‑
茚的合成
[0250][0251]
将one(主要基于如下化合物的混合物:2,3
‑
二氢
‑6‑
(2
‑
羟基
‑2‑
甲基
‑1‑
氧代丙基)
‑
1,1,3
‑
三甲基
‑3‑
[4
‑
(2
‑
羟基
‑2‑
甲基
‑1‑
氧代丙基)苯基]
‑
1h
‑
茚;2,3
‑
二氢
‑5‑
(2
‑
羟基
‑2‑
甲基
‑1‑
氧代丙基)
‑
1,1,3
‑
三甲基
‑3‑
[4
‑
(2
‑
羟基
‑2‑
甲基
‑1‑
氧代丙基)苯基]
‑
1h
‑
茚)(0.2g,0.489mmol)和氯五甲基二硅烷(0.16g,0.977mmol)溶解在10ml二氯甲烷中,加入0.07g(0.977mmol)咪唑。将反应混合物在室温搅拌3小时,然后真空蒸发。粗产物用20ml甲苯中稀释,经硅胶过滤。真空除去溶剂,得到纯产物。
[0252]1h nmr(cdcl3,δppm):0.00(m,18h),0.12(m,18h),1.1(s,3h),1.39(s,3h),1.59(m,12h),1.73(s,3h),2.25(d,1h),2.49(m,1h),7.1
‑
7.22(m,3h),7.93
‑
8.25(m,4h)。
[0253]
实施例12
[0254]1‑
(9
‑
丁基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑2‑
[(1,1,2,2,2
‑
五甲基二硅烷
‑1‑
基)氧基]丙
‑1‑
酮的合成
[0255][0256]
将n
‑
丁基咔唑(3g,13.4mmol)和α
‑
溴代异丁酰溴(3.67g,16mmol)溶于40ml苯中,进一步在0℃和搅拌条件下于30分钟内加入氯化铝(1.78g,13.4mmol)。添加完成后,在25℃
下搅拌4小时。将反应溶液倒入50g冰水中,再用100ml苯萃取。有机层用硫酸镁干燥,过滤移除干燥剂,然后通过硅胶柱色谱法(甲苯/石油醚6:1)纯化残留物。将粗产物溶解在6ml二氯甲烷中,在回流条件下用0.24g 50%naoh溶液处理5小时。用10ml水和10ml二氯甲烷稀释后,分离两相。有机相用水洗涤,用硫酸钠干燥,过滤并真空蒸发溶剂。粗产物通过快速柱色谱法纯化(二氯甲烷/乙酸乙酯9:1),用于下一步骤。
[0257]
将1
‑
(9
‑
丁基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
羟基
‑2‑
甲基丙
‑1‑
酮(0.5g,1.61mmol)和氯五甲基二硅烷(0.26g,1.61mmol)溶解在5ml二氯甲烷中,加入0.1g(1.61mmol)咪唑。反应混合物在30℃搅拌3小时,然后减压蒸发。粗产物通过快速柱色谱(甲苯)纯化。真空除去溶剂,得到纯产物(0.3克)。
[0258]1h nmr(cdcl3,δppm):0.00(s,9h),0.18(s,6h),0.92(t,3h),1.4(m,2h),1.65(s,6h),1.85(m,2h),4.32(t,2h)7.2
‑
7.5(m,4h),8.14(d,1h),8.40(d,1h),9.14(s,1h)。
[0259]
实施例13
[0260]
比较试验
[0261]
本发明的α
‑
二甲硅烷氧基酮与根据macromolecules 2009,42,6031
‑
6037(pi
‑
1)的描述制备的现有技术的2
‑
甲基
‑2‑
(1,1,2,2,2
‑
五甲基二甲硅烷氧基)
‑1‑
苯基丙
‑1‑
酮(comp
‑
1)和2
‑
羟基
‑2‑
甲基
‑1‑
苯基丙酮(comp
‑
2)进行比较。
[0262]
实施例13.1
[0263]
比较试验
[0264]
实施例13.1.1.
[0265]
透明制剂水银灯
[0266]
将光引发剂以3重量%的浓度各自溶解在由605和350(allnex)组成的99.5:0.5(重量)混合物中,制备用于测试的可光聚合组合物。
[0267]
将放置在ft
‑
ir(ft
‑
ir 430
‑
jasco)样品舱中的可光聚合组合物暴露于水银灯(160w),与样品的距离为65mm,角度为30
°
。在光聚合过程中每隔恒定的时间间隔获取ir光谱,使用ir软件确定丙烯酸双键的峰面积在1408cm
‑1和810cm
‑1处随时间而减少。该数据量化了聚合度,从而量化了光引发剂的效率。
[0268]
表示为随时间的聚合度百分比的结果在表3中列出。
[0269]
表3.
[0270][0271]
*比较实施例
[0272]
这些结果证实,在以相同的数量使用时,式(ia)和(ii)所示的化合物具有与比较实施例(comp
‑
1)相当或更佳的反应性。实施例5的化合物达到了最佳性能,该化合物的反应性是参考化合物(comp
‑
1)的2倍。
[0273]
实施例13.1.2.
[0274]
透明制剂led灯(400nm)
[0275]
按照以下方法制备测试用的可光聚合的组合物:
[0276]
1.将浓度为3%(重量)的每种光引发剂与0.5%异丙基噻吨酮(敏化剂)溶解在由605和350(allnex)组成的99.5:0.5混合物中(制剂a)。
[0277]
2.将浓度为3%(重量)的每种光引发剂溶解在由605和350(allnex)组成的99.5:0.5混合物中(制剂b)。
[0278]
将放置在ft
‑
ir(ft
‑
ir 430
‑
jasco)样品舱中的可光聚合组合物暴露于led光源(400nm),该光源与样品的距离为25mm,角度为30
°
。在光聚合过程中每隔恒定的时间间隔获取ir光谱,使用ir软件确定丙烯酸双键的峰面积在1408cm
‑1和810cm
‑1处随时间而减少。
[0279]
该数据量化了聚合度,从而量化了光引发剂的效率。
[0280]
表示为随时间的聚合度百分比的结果在表4中列出。
[0281]
表4
[0282][0283]
*比较实施例
[0284]
这些结果证实,式(i)、(ia)和(ii)所示的化合物能够被敏化并且能够与led灯一起工作,而这对于比较实施例是α
‑
羟基酮(comp
‑
2)来说是无法实现的。
[0285]
令人惊讶地发现,所有接受测试的α
‑
二甲硅烷氧基芳基酮当与敏化剂混合时都能够与led光源一起工作。
[0286]
实施例13.1.3.
[0287]
青色喷墨油墨
[0288]
将放置在ft
‑
ir(ft
‑
ir 430
‑
jasco)样品舱中的可光聚合组合物暴露于水银灯(160w),该光源与样品的距离为65mm,角度为30
°
。在光聚合过程中每隔恒定的时间间隔获取ir光谱,使用ir软件确定丙烯酸双键的峰面积在1408cm
‑1和810cm
‑1处随时间而减少。该数据量化了聚合度,从而量化了光引发剂的效率。
[0289]
表示为随时间的聚合度百分比的结果在表5中列出。
[0290]
表5
[0291][0292]
*比较实施例
[0293]
这些试验证实,式(ia)的化合物相对于比较实施例具有更高的反应性。
[0294]
实施例13.1.4.
[0295]
青色喷墨油墨led灯(400nm)
[0296]
按照以下方法制备测试用的可光聚合的组合物:
[0297]
1.将浓度为5%(重量)的每种光引发剂与0.5%异丙基噻吨酮(敏化剂)溶解在青色喷墨油墨中(制剂a)。
[0298]
2.将浓度为5%(重量)的每种光引发剂溶解在青色喷墨油墨中(制剂b)。
[0299]
将放置在ft
‑
ir(ft
‑
ir 430
‑
jasco)样品舱中的可光聚合组合物暴露于led光源(400nm),该光源与样品的距离为25mm,角度为30
°
。在光聚合过程中每隔恒定的时间间隔获取ir光谱,使用ir软件确定丙烯酸双键的峰面积在1408cm
‑1和810cm
‑1处随时间而减少。
[0300]
该数据量化了聚合度,从而量化了光引发剂的效率。
[0301]
表示为随时间的聚合度百分比的结果在表6中列出。
[0302]
表6
[0303][0304]
*比较实施例
[0305]
这些结果证实,式(i)所示的化合物能够在着色系统中被敏化,所以它们能够与led灯一起工作,而这对于比较实施例(comp
‑
2)来说是无法实现的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。