1.本发明涉及分子生物学技术领域,尤其涉及毕赤酵母中新型生长依赖型启动子及其应用。
背景技术:
2.毕赤酵母(pichia pastoris)具有生长快、可高密度发酵等优势,除了常被用作蛋白表达宿主外(biotechnology journal,2019,14(6):1800694),在小分子化合物生物合成方面也表现出了良好的应用潜力(microbial cell factories,2013,12(1):1
‑
14;applied and environmental microbiology,2005,71(7):3453
‑
3457)。相比于酿酒酵母,有关毕赤酵母的基础研究远远落后,导致其中可用的启动子元件严重不足,限制了毕赤酵母的工程化应用。
3.目前在毕赤酵母中常用的强启动子分为2类:组成型强启动子和诱导型强启动子。组成型强启动子是指在该类启动子控制下,基因的表达具有持续性且恒定在较高的水平,不受诱导物或者抑制物的影响。诱导型强启动子是指该类启动子在常规条件下不发挥作用,只有在特定的信号刺激下,才大幅度地提高下游基因的转录水平,实现蛋白的特异性表达。
4.目前,基于毕赤酵母进行目标蛋白表达时,通常使用来源于其甲醇利用(mut)途径的诱导型强启动子,包括p
aox1
、p
das
、p
fld1
(nucleic acids research,1987,15(9):3859
‑
76;gene,1998,216(1):93
‑
102)等。由于甲醇易燃且具有细胞毒性,在一定程度上限制了甲醇诱导型强启动子在毕赤酵母中的应用。在以葡萄糖、甘油等为主要碳源的培养基中进行毕赤酵母发酵时,常会使用不依赖于甲醇的组成型强启动子介导目标蛋白的表达,例如p
gap
(gene,1997,186(1):37
‑
44)、p
tef1
(applied microbiology and biotechnology,2007,74(3):601
‑
608)、p
gcw14
(biotechnology letters,2013,35(11):1865
‑
1871)等。由于组成型启动子活性强,不具有时间特异性表达模式,在表达具有毒性的目标蛋白时,会对酵母细胞的生长产生抑制,具有一定的局限性。因此,发掘和鉴定兼具上述两类启动子优势的毕赤酵母内源性强启动子,具有重要的理论意义和应用价值。
技术实现要素:
5.本发明的目的在于针对现有技术的不足,提供一种毕赤酵母中新型生长依赖型启动子及其应用。
6.本发明的目的是通过以下技术方案来实现的:一种毕赤酵母来源的生长依赖型启动子,其碱基序列如seq id no.1或seq id no.2所示。
7.一种上述毕赤酵母来源的生长依赖型启动子在表达盒中的应用,所述表达盒除包含上述的任一启动子外,还包含其下游连接的结构基因或调节基因,以及终止子。
8.进一步地,所述终止子优选但不限于adh1终止子、aox1终止子。
9.进一步地,所述下游连接的结构基因或调节基因包括但不限于β
‑
半乳糖苷酶基因
lacz、粒细胞
‑
巨噬细胞集落刺激因子基因csf2*。
10.本发明所述的具有生长依赖型启动子活性的dna序列共有500个碱基,来源于毕赤酵母(pichia pastoris),seq id no.1位于基因pas_chr2
‑
2_0208(ncbi gene id:8198616、locus:xp_002491989)起始密码子atg前的5’端非编码区。seq id no.2位于基因pas_chr4_0627(ncbi gene id:8201336、locus:xp_002494066)起始密码子atg前的5’端非编码区。为了便于后续叙述的方便,在本发明中将上述启动子分别命名为p
0208
、p
0627
,对应的基因分别命名为0208、0627。
11.本发明所述dna可用于构建毕赤酵母表达载体,通过将载体整合入毕赤酵母基因组,可在毕赤酵母中表达由该启动子控制的目标蛋白。
12.本发明分别扩增用于将表达载体整合到酵母基因组上的插入位点序列(seq id no.3)、复制子序列(seq id no.4)、抗性基因的表达盒序列(seq id no.5)、目标基因lacz或csf2*的编码序列(seq id no.6或seq id no.7)、终止子序列(seq id no.8)、组成型强启动子p
gap
的dna序列(seq id no.9)或本发明提及的生长依赖型启动子p
0208
的dna序列(seq id no.1)或p
0627
的dna序列(seq id no.2)。将上述各序列的dna片段以无缝克隆的方式连接获得表达载体。其中目标基因可以但不限于lacz、csf2*,任何功能基因均可以替换它们,被本发明所述的dna片段所驱动表达。
13.所述毕赤酵母表达载体经酶切线性化后,电转入毕赤酵母gs115 wt菌株(invitrogen
tm
)中。将在相应的抗性平板上长出的转化子进行液体培养后,抽取基因组进行pcr验证,筛选出含有表达载体的毕赤酵母菌株。
14.本发明的有益效果是:在分别以葡萄糖、甘油为碳源的培养基中,该启动子均具有类似的特征:即随着细胞的生长,该启动子控制下的基因在酵母对数生长期转录水平低,稳定期转录水平高。该启动子具有的生长依赖表达特性可以将毕赤酵母细胞的生长和蛋白的表达分隔开,对毒性蛋白的异源表达具有良好的应用潜力。
附图说明
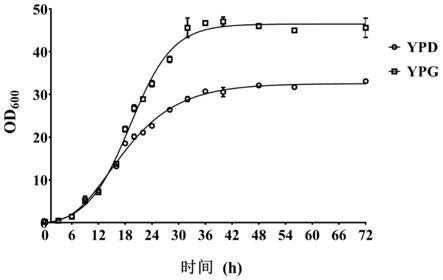
15.图1为毕赤酵母gs115 wt菌株在ypd、ypg培养基中的生长曲线;
16.图2为毕赤酵母gs115 wt菌株在ypd、ypg培养基中分别培养16h、36h、60h时基因gap、0208和0627的相对转录水平柱状图(act1为内参基因);
17.图3为构建的酵母载体ppic
‑
lacz图谱;
18.图4为利用本发明的启动子p
0208
、p
0627
构建的酵母表达载体p0208
‑
lacz、p0627
‑
lacz图谱;
19.图5为在50ml ypd培养基中、不同生长阶段,由本发明启动子p
0208
、p
0627
控制的lacz的相对转录水平变化柱状图(act1为内参基因);
20.图6为在50ml ypd培养基中、不同生长阶段,由本发明启动子p
0208
、p
0627
控制的β
‑
半乳糖苷酶的活性变化以及相关菌株的生物量(ood
600
)变化柱状图;
21.图7为在商品化载体ppic9基础上构建的酵母表达载体ppic9
‑
csf2*图谱;
22.图8为利用本发明启动子所构建的酵母表达载体p0208
‑
csf2*、p0627
‑
csf2*图谱以及利用组成型强启动子p
gap
构建的酵母表达载体pgap
‑
csf2*图谱;
23.图9为利用本发明启动子驱动表达csf2 t85a
的western blot检测结果图,图中,孔
道1为空白对照,孔道2为本发明启动子p
0208
驱动表达的csf2 t85a
,孔道3为本发明启动子p
0627
驱动表达的csf2 t85a
,孔道4为阳性对照,组成型强启动子p
gap
驱动表达的csf2 t85a
,孔道5为marker。
具体实施方式
24.下面结合附图并通过具体实施方式来进一步说明本发明的技术方案,但下述的实例仅仅是本发明的简易例子,并不代表或限制本发明的权利保护范围,本发明的保护范围以权利要求书为准。
25.以下实施例中,若无特殊说明,所有的试剂及耗材均购自本领域常规试剂厂商;若无特殊说明,所用的实验方法和技术手段均为本领域常规的方法和手段,质粒的抽提、rna的获取、dna片段的回收、连接等操作详见相应试剂盒的说明书。培养基的配置、酵母细胞的培养等详见invitrogen
tm
公司的操作手册“user guide:pichia expression kit”(来源:https://www.thermofisher.cn/order/catalog/product/k171001#/k171001)。毕赤酵母gs115 wt菌株、载体ppiczαc和ppic9均购买于invitrogen
tm
公司;酿酒酵母by4741 wt菌株和大肠杆菌bl21(de3)wt菌株、dh5αwt菌株均购买于北京华越洋生物公司。将毕赤酵母gs115 wt菌株作为基础,将lacz或者csf2*导入该毕赤酵母中产生一系列的菌株均是为了评估本发明启动子的活性,在此过程中获得的菌株并不属于本专利所保护的范畴。
26.实施例1:本发明启动子特性的初步评价
27.为了检测本发明启动子的特性,分别在ypd、ypg两种液体培养基中对gs115 wt菌株进行培养,检测在酵母的不同生长阶段,本发明启动子p
0208
、p
0627
对应基因0208、0627的转录水平。具体操作如下:
28.(1)生长曲线的测定
29.a.挑选健壮的gs115 wt单克隆于5ml ypd液体培养基中过夜培养。
30.b.取适量上述培养液分别转接到50ml ypd、ypg液体培养基中,使初始od
600
~0.2,30℃、220rpm连续培养72h,间隔取样,绘制生长曲线。
31.结果:毕赤酵母gs115 wt菌株在两种培养基中均需生长约36h后,才能进入稳定期。尽管两种培养基碳原子的摩尔数类似(10.86mmol c3h8o
3 vs.5.56mmol c6h
12
o6),但酵母在50ml ypg培养基中的生物量(od
600
~46)高于其在50ml ypd培养基中的生物量(od
600
~33),具体结果见图1。
32.(2)0208、0627基因转录水平的检测
33.a.根据上述生长曲线,选取16h、36h、60h作为取样点,在两种培养基中,分别收取总细胞量为od
600
~16的酵母细胞。5000
×
g离心3min,去上清。
34.b.参照酵母rna抽提试剂盒(omega bio
‑
tek)说明书,提取酵母细胞样品总rna,利用1%(w/v)琼脂糖凝胶电泳检测所提取的总rna完整性,利用超微量分光光度计(nanodrop 2000)读取总rna的浓度。
35.c.利用dna核酸内切酶dnaseⅰ(takara)处理总rna中残余的基因组。rna用量为10μg,反应体系为50μl,dnaseⅰ用量为4μl。37℃反应40min后,再次利用酵母rna抽提试剂盒回收纯净的rna。
36.d.利用反转录酶m
‑
mlv rnase h
‑
(takara),反转录1μg rna,获得cdna。
37.e.利用tbpremix ex taq
tm
(takara)进行rt
‑
qpcr的分析,扩增片段的大小为150bp左右,引物设置在基因orf的近3’端。
38.f.以act1(基因id:pas_chr3_1169)为内参基因,通过δct值计算0208、0627相对内参基因的转录水平。rt
‑
qpcr所用引物序列见表1。
39.表1实施例1中rt
‑
qpcr所用的序列
40.引物名称序列(5
’‑3’
)0208 rt f/rgcacggcagatacacttgct/cggtgtatcgctcgccactt0627 rt f/rcaaggctgccaacaaattgac/ttaccagcttccaatgcatcaccgap rt f/rgtttggctttccgtgtccca/cggcatcttcagtgtaacccact1 rt f/rtgcaaaaggagcttactgcc/tggtccagattcgtcgtact
41.结果:本发明启动子控制的原生基因0208、0627的相对转录水平在两种培养基中均表现为:在对数生长期转录水平很低,在稳定期转录水平显著提高,即随着酵母细胞的生长,二者转录水平呈递增的趋势。以gap(pas_chr2
‑
1_0437,甘油醛
‑3‑
磷酸脱氢酶基因,具有较高的转录水平)为阳性对照,在ypd、ypg培养基中,当酵母进入生长稳定期后,0208、0627的相对转录水平均高于gap的相对转录水平,暗示在稳定期本发明启动子p
0208
、p
0627
的活性均高于p
gap
的活性,因此本发明启动子表现出良好的应用潜力。具体结果见图2。
42.实施例2:表达载体p0208
‑
lacz、p0627
‑
lacz的构建
43.(1)利用质粒小提试剂盒(诺唯赞)提取质粒ppiczαc。
44.(2)毕赤酵母gs115 wt菌株、酿酒酵母by4741 wt菌株、大肠杆菌bl21(de3)wt菌株基因组的提取:
45.收取1ml培养过夜的上述细胞(酵母使用ypd培养基培养,大肠杆菌使用lb培养基培养),高速离心后收集沉淀,用200μl裂解液、200μl苯酚/氯仿/异戊醇的混合溶液(25:24:1)、200μl 1
×
te重悬,加入玻璃砂震荡裂解细胞,经高速离心后,回收最上层的液体,加入2.5倍体积的无水乙醇,低温条件下对基因组沉降1h以上。高速离心后,用70%(v/v)乙醇洗涤,获得纯净的基因组。用200μl1
×
te重悬基因组,分装备用。
46.(3)以ppiczαc为模板,使用高保真酶hieff canace(翊圣)分别扩增用于载体整合到酵母基因组上的插入位点p
aox1
序列(seq id no.3)、复制子序列(seq id no.4)、以及博来霉素抗性基因的表达盒序列(seq id no.5);以大肠杆菌bl21(de3)wt菌株的基因组为模板,扩增lacz的编码序列(seq id no.6)。以酿酒酵母by4741 wt菌株的基因组为模板,扩增终止子t
adh1
序列(seq id no.8)。上述片段经过1%(w/v)琼脂糖凝胶电泳进行分离,使用凝胶dna小量回收试剂盒(诺唯赞)进行纯化。
47.(4)利用无缝克隆试剂盒(全式金)将上述各个片段(对应序列为seq id no.3~6、8)组装成载体ppic
‑
lacz(图3)。
48.(5)以毕赤酵母gs115 wt菌株的基因组为模板,使用高保真酶hieff canace分别扩增启动子p
0208
序列(seq id no.1)、p
0627
序列(seq id no.2)。利用限制性内切酶hindⅲ和stuⅰ处理载体ppic
‑
lacz,37℃酶切2~3h,酶切产物经过琼脂糖凝胶电泳进行分离,使用凝胶dna小量回收试剂盒进行纯化。利用无缝克隆试剂盒将p
0208
序列、p
0627
序列分别与酶处理后的载体ppic
‑
lacz进行反应(50℃反应15min后转化大肠杆菌dh5α),获得表达载体p0208
‑
lacz、p0627
‑
lacz(载体图谱见图4)。
49.实施例3:利用lacz报告系统评估本发明的启动子活性
50.来源于大肠杆菌的β
‑
半乳糖苷酶可以将无色的2
‑
硝基苯
‑
β
‑
d
‑
半乳糖苷(onpg)水解成半乳糖和黄色的邻硝基苯酚,因此可通过比色方法检测酶活。在酵母中,β
‑
半乳糖苷酶基因lacz常被用于构建启动子报告系统,通过在该基因上游连接目标启动子来评估启动子的转录活性。
51.具体操作如下:
52.(1)利用质粒小提试剂盒(诺唯赞)提取载体p0208
‑
lacz、p0627
‑
lacz。
53.(2)利用限制性内切酶pmeⅰ在p
aox1
序列内部进行酶切,37℃处理2~3h,分别将上述两个载体线性化,以p
aox1
序列作为同源重组片段,通过电转的方式,将载体整合到gs115 wt菌株的基因组中。
54.(3)电击结束后立即加入1ml预冷的1m山梨醇,30℃孵育1h后,分别将含有p0208
‑
lacz或p0627
‑
lacz载体的菌液涂布到博来霉素浓度为100μg/ml的ypd固体平板上,培养48h,待转化子长出。
55.(4)随机挑选转化子于5ml ypd培养基中过夜培养,提取各个转化子的基因组,利用pcr进行鉴定,成功筛选到由本发明启动子控制的、可以表达β
‑
半乳糖苷酶的酵母菌株gs115/p0208
‑
lacz、gs115/p0627
‑
lacz。
56.这些菌株的起始菌株均为gs115 wt菌株,它们只是用来评估本发明启动子的活性,任何由本发明启动子控制的目标基因均可以在gs115 wt菌株中表达,该方法具有通用性,菌株并不是本专利所保护的范畴。
57.(5)利用rt
‑
qpcr评估本发明启动子的特性。
58.以50ml ypd培养基为例,基因组中分别含有p0208
‑
lacz、p0627
‑
lacz的各个酵母菌株的初始接种量为od
600
~0.2,30℃、220rpm连续培养60h,每12h进行取样后抽提rna,检测由p
0208
、p
0627
控制的lacz的相对转录水平。rna的抽提、残余基因组的去除以及反转录的方法详见实施例1。rt
‑
qpcr所用的引物见表2(act1 rt引物见表1)。
59.表2实施例3中rt
‑
qpcr所用的引物
60.引物名称序列(5
’‑3’
)lacz rt f/rgcacggcagatacacttgct/cggtgtatcgctcgccactt
61.结果:在ypd培养基中(起始od
600
~0.2)培养12h~60h后,lacz的相对转录水平表明,p
0208
、p
0627
的活性在细胞对数生长期很低,随着细胞的生长,二者活性逐步增强,表现出生长依赖的特性,且p
0627
活性强于p
0208
(图5)。
62.(6)lacz编码的β
‑
半乳糖苷酶常作为报告蛋白,其编码序列置于待评估的启动子后,利用该酶的活性表征对应启动子的强度。
63.a.在平板上挑选健壮的gs115 wt单克隆于5ml ypd培养基中过夜培养。
64.b.取适量上述培养液分别转接到50ml ypd、ypg液体培养基中,使初始od
600
~0.2,30℃、220rpm培养60h。
65.c.选择不同生长时期的酵母细胞进行分析,方法参照文献journal of bioscience&bioengineering,2017:s1389172317303250。具体如下:选取适量的细胞,5000
×
g离心3min,取沉淀用z buffer(10mm氯化钾、1mm硫酸镁、60mm磷酸氢二钠以及40mm磷酸二氢钠)重悬,测定od
600
(此时od
600
应介于02
‑
0.8范围内,此值即为下面公式中的od
600
)。d.
取适量体积(下面公式中的v)的上述稀释液和z buffer混合,使最终的体积为1ml,加入50μl氯仿和25μl 0.1%(w/v)sds,涡旋30s,置于30℃的水浴锅中反应15min,使细胞内容物释放。
66.e.加入200μl 4mg/ml的onpg反应液,开始计时,待eppendorf管中的反应液的颜色变成淡黄色时,立即加入500μl 1m na2co3并混匀,结束计时(下面公式中的t)。
67.f.上述液体经12000
×
g离心5min,取上清液检测od
420
和od
550
。
68.g.根据以下公式计算β
‑
半乳糖苷酶的酶活:
69.miller units=1000
×
(od
420
‑
1.75
×
od
550
)/(v
×
od
600
×
t)
70.其中v为反应时加入的菌液的体积(ml),t为加入onpg后的反应时间(min)。
71.结果:在不同生长阶段、两种培养基中,表达lacz的菌株中由本发明启动子控制的β
‑
半乳糖苷酶活性均表现出在对数生长期较低、稳定期显著增强的特性,与转录水平的变化相一致。另外,对应菌株的生长无明显差异(图6)。
72.实施例4:利用分泌蛋白csf2 t85a
评估本发明的启动子的活性
73.由于毕赤酵母具有良好的蛋白分泌系统,且胞外蛋白较少,利用其表达的目标蛋白一般采用分泌表达方式,以便于后续的分离与纯化。为了进一步评估本发明启动子的实用性,用本发明启动子控制小鼠来源的粒细胞
‑
巨噬细胞集落刺激因子2(csf2)蛋白突变体csf2 t85a
的表达,并与组成型强启动子p
gap
控制的蛋白表达进行比较。具体操作如下:
74.(1)载体p0208
‑
csf2*、p0627
‑
csf2*、pgap
‑
csf2*的构建
75.a.以ppic9作为原始载体,将截去前17个氨基酸编码序列(预测为信号肽)的目标基因csf2进行密码子优化并在序列3’端终止密码子前添加6
×
his标签,人工合成后插入α
‑
分泌肽后,与之融合表达。由于该蛋白具有两个n
‑
糖基化修饰位点,为方便研究,将第一个n
‑
糖基化修饰位点进行突变(t85a),突变后的核酸序列(含有6
×
his标签)命名为csf2*(序列见seq id no.7),编码的蛋白命名为csf2 t85a
,最终获得载体ppic9
‑
csf2*(载体图谱见图7,使用的启动子为甲醇诱导型启动子p
aox1
)。该过程由上海捷瑞生物工程有限公司完成。
76.b.选用限制性内切酶bamhⅰ和ndeⅰ处理载体ppic9
‑
csf2*,37℃酶切2~3h,回收含有csf2*的载体片段。以ppic9
‑
csf2*载体为模板,使用hieff canace高保真dna聚合酶扩增含有复制子和抗性基因表达盒的dna序列,以gs115 wt基因组为模板扩增本发明启动子的dna序列以及组成型强启动子p
gap
的dna序列,利用无缝克隆的方式获得载体p0208
‑
csf2*、p0627
‑
csf2*、pgap
‑
csf2*(载体图谱见图8,三个载体只有启动子不同,其他序列均相同)。(2)将目标基因csf2*导入毕赤酵母中
77.a.分别将2μg ppic9
‑
csf2*、p0208
‑
csf2*、pgap
‑
csf2*通过限制性内切酶salⅰ进行线性化处理。
78.b.参照实施例3中的方法,将上述载体分别电转入毕赤酵母gs115 wt菌株中,加入山梨醇后,30℃孵育1h,分别取200μl含有上述载体的菌液涂布到md平板上,30℃培养48h,待转化子长出。
79.c.随机挑选转化子于5ml ypd培养基中过夜培养,利用实施例2中提取基因组的方法获得各个转化子的基因组,并进行pcr验证,成功筛选到由本发明启动子控制的、可以表达csf2
t85a
的酵母菌株gs115/p0208
‑
csf2*、gs115/p0627
‑
csf2*以及由组成型强启动子控制的、可以表达csf2 t85a
的酵母菌株gs115/pgap
‑
csf2*。
80.这些菌株的起始菌株均为gs115 wt菌株,它们只是用来评估本发明启动子的活性,任何由本发明启动子控制的目标基因均可以在gs115 wt菌株中表达,该方法具有通用性,菌株并不是本专利所保护的范畴。
81.(3)由本发明启动子控制的csf2 t85a
的表达及纯化
82.a.酵母菌株gs115/p0208
‑
csf2*、gs115/p0627
‑
csf2*、gs115/pgap
‑
csf2*分别在50mlbmgy培养基中30℃,220rpm培养(起始od
600
~0.2)。
83.b.培养48h后,培养液分别经离心去除沉淀,向上清液中缓慢加入(nh4)2so4,使其饱和度达到80%(w/v),放置于4℃沉降过夜,12000
×
g,4℃离心20min,去除上清液。
84.c.沉淀分别用20mm tris
‑
hcl(ph7.5)悬浮,加入100μl ni
‑
nta镍离子柱(纽龙生物),密封后置于冰上,在水平摇床上孵育2
‑
3h,使蛋白可以和ni
‑
nta充分结合。最后将结合液沿管壁缓慢加入亲和层析柱(生工)中,静置5min后,释放流出液。
85.d.待所有液体基本流出后,分别向柱中分3次加入2ml 20mm咪唑缓冲液,收集洗脱液并做好标记。随后,向柱中分3次加入0.5ml 250mm咪唑缓冲液,收集洗脱液并做好标记。(4)由本发明启动子控制表达的目标蛋白csf2 t85a
的western blot检测
86.a.配置15%(w/v)分离胶和5%(w/v)浓缩胶。
87.b.向电泳槽中加入1
×
sds电泳缓冲液,并没过加样胶孔。取等体积的250mm咪唑洗脱液(分别含有不同启动子控制表达的csf2 t85a
),加入1
×
上样缓冲液,沸水浴中放置5min,冷却至室温后微离心,上清液全部加入加样孔中。
88.c.将电压设置为100v,开始电泳,待彩色预染蛋白marker(诺唯赞)出现分离后,将电压调整为120v,继续电泳,当marker条带完全分开后终止电泳。
89.d.将含有目标蛋白条带的胶块切割下来,剪一块略大于胶块的硝酸纤维素膜置于1
×
转膜液中浸泡,按照负极
‑
海绵
‑
3层滤纸
‑
胶
‑
硝酸纤维素膜
‑
3层滤纸
‑
海绵
‑
正极(避免出现气泡)的顺序将转膜夹放入转膜槽中,转膜槽置于冰水混合物中,电压设置为100v、时间设置为60min。
90.e.转膜结束后取出硝酸纤维素膜,置于5%(w/v)的脱脂奶粉中封闭1h。倒掉封闭液,用1
×
tbst洗膜,每次5min,洗6次。
91.f.用1
×
tbst将小鼠抗6
×
his单克隆抗体(生工)按1:3000的比例稀释后,均匀覆盖到硝酸纤维素膜上,在水平摇床上孵育1h。将膜取出后,用1
×
tbst洗膜,每次5min,洗6次。
92.g.用1
×
tbst将辣根过氧化物酶标记的山羊抗小鼠igg(生工)按1:3000的比例稀释后,均匀覆盖到硝酸纤维素膜上,室温下孵育1h。将膜取出后,用1
×
tbst洗膜,每次5min,洗6次。
93.h.将高灵敏ecl发光试剂(生工)均匀覆盖到硝酸纤维素膜上,进行反应。将膜置于化学发光成像仪中进行曝光,观察条带并拍照。
94.结果:如图9所示,本发明的启动子可以介导蛋白csf2 t85a
的表达,p
0627
效果优于p
0208
,与组成型强启动子p
gap
介导的蛋白表达具有可比性,且不依赖于甲醇诱导,在蛋白表达方面表现出较大的潜力。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。