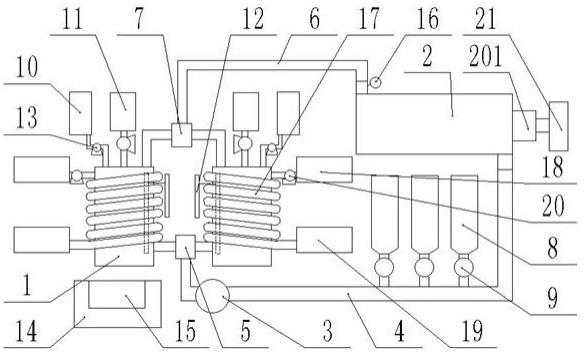
1.本发明属于催化剂制备领域,具体涉及一种在低温下由丙烷直接脱氢高效合成丙烯的催化剂及制备方法和应用。
背景技术:
2.随着石油资源日益短缺,价格与日攀升,未来世界经济发展面临能源、资源短缺限制的同时,也促进了世界能源格局的改变。而页岩气作为一种新型的非常规天然气资源正被大量开发,以其储量大、开采寿命长和生产周期长等优点著称。近年来,通过改进水力压裂技术来有效提取页岩气的手段获得不断发展,有效地替代了丙烷的来源,而传统的丙烯生产工艺的产能增长和开工率由于疫情等的影响,受到严重的压缩。因此,如何以有效的方式将页岩气转化成高附加值化学品,尤其是将其应用在丙烷脱氢制取高价值丙烯方面显示出良好的经济效益。
3.丙烯作为最重要的生产结构单元之一,可用于生产众多重要的有机化工原料,如:聚丙烯、丙烯晴、丁/辛醇、环氧丙烷、苯酚/丙酮、丙烯酸及酯、环氧氯丙烷等;生产精细化学品,如:合成树脂、合成纤维、合成橡胶等。此外,在炼油工业上也可用作原料用来生产叠合汽油,减缓能源危机。另外,还可应用于环保、医学科学和基础研究等领域。
4.迄今为止,能够实现丙烷脱氢工业化的催化剂仍只有ptsn/al2o3或者 cro
x
/al2o3催化剂。因此,近几十年来,广泛的研究主要集中在pt基和cro
x
基催化剂的改性、减少用量、以及设计催化剂结构来提高催化效率、降低成本和更加环保、延长反应寿命。因此,开发一种可替代的低成本可控的丙烷脱氢催化剂对于天然气资源的利用仍是十分必要。
5.由于丙烷直接脱氢过程具有较高的吸热率(
∆
hθ298 = 124.3 kj
·
mol
‑1),需要较高的反应温度(550
‑
700 ℃)。这种苛刻的反应条件对催化剂的热稳定性构成了挑战。而在较低温度下实现丙烷脱氢具有降低能耗、实现绿色化学、抑制结焦和烧结等一系列优点,引起了越来越多的研究者们的兴趣。考虑到丙烷直接脱氢的热力学平衡限制,如何实现低温下具有良好催化性能的丙烷直接脱氢过程是一个新兴的重大挑战。因此,开发具有良好低温催化性能的高稳定性催化剂是十分迫切的。
6.在众多最有前途的丙烷直接脱氢反应催化剂中,单原子pt催化剂因其具有明确、孤立的活性位点、独特而优越的催化性能等优势而得到了广泛的研究和应用。在低温条件下,原子分散的pt位点可以很容易地解离烷烃的第一和第二c
‑
h键,有效地减少了生成焦炭的副反应。但孤立的pt活性位点的表面吉布斯自由能高,催化过程中会发生团聚和烧结,进而导致催化剂失活。为合理设计pdh催化剂,提出了许多策略。例如,探索了过渡后金属元素(如sn、zn、ga、in)作为共催化剂,以改善pt的分散性、几何结构和电子性能,提高活性、选择性、稳定性和抗结焦性能。而氧化铝对氧化镓有较强的载体效应,进而有助于维持pt物种的分散。另一方面,通过在催化剂上表面包裹无机氧化物壳层作为物理屏障,防止pt物种的聚集,可以提高催化剂的热稳定性和抗结焦性能。cn112135687a公开了一种浸渍法负载k掺杂的ptga/al2o3‑
sio2复合催化剂,该催
化剂用于固定床反应器,在5 bar压力下,570 ℃反应温度,10%的丙烷,空速12h
‑1的反应条件下,引入5 wt% sio2的催化剂为性能最佳的催化剂,初始1分钟时转化率为~41%,11分钟后降为~40%。和纯al2o3做载体的催化剂对比,不同sio2引入量的催化剂稳定性先随着sio2的增加而提高,同时引入的b酸位点也导致副反应更加容易发生。因此存在一个最佳的sio2引入量范围,但由于副反应导致的失活现象仍然存在。
7.cn112221493a公开了一种制备微量贵金属修饰的均匀分布pt
‑
ga2o3/al2o3和rh
‑
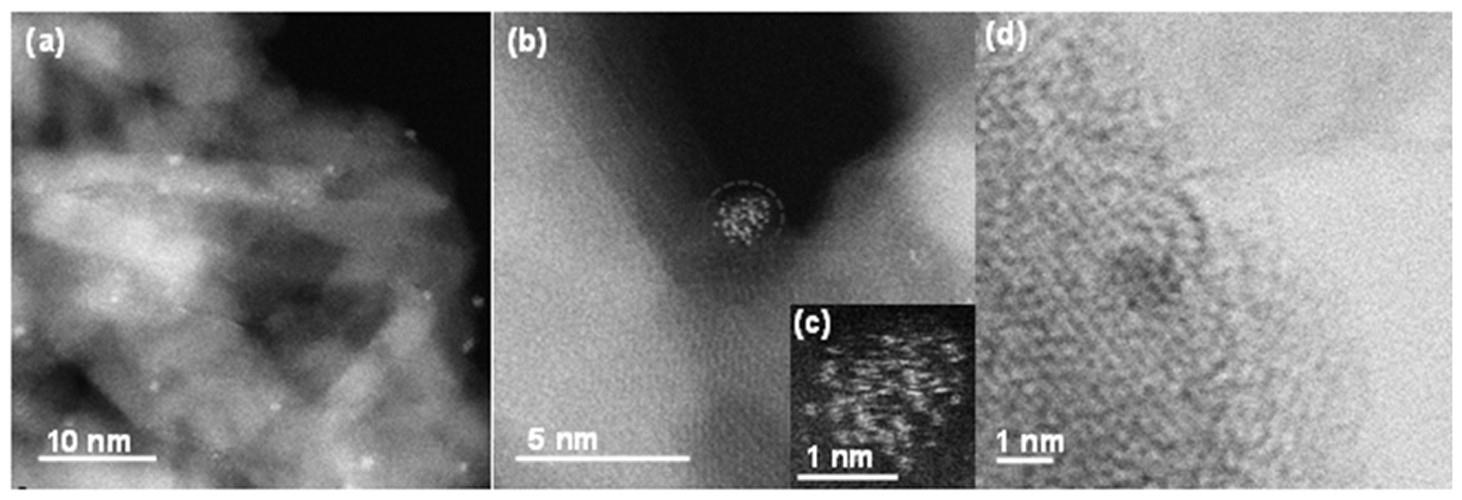
ga2o3/al2o3催化剂,负载活性组分为ga2o3,微量贵金属为助剂,在反应压力0.1 mpa,反应温度600
ꢀ°
c,c3h8/h
2 =1/1,氮气作为平衡气,质量空速10 h
‑1的条件下,不同pt负载量的催化剂初始转化率为19%~27%,反应4h后为8%~17%,选择性基本维持88~99%左右,显示出快速的失活性质。
8.cn110237840b公开了一种采用静电吸附法制备单原子pt/tio2催化剂,反应压力为常压,反应温度580
ꢀ°
c,c3h8/h
2 =1/1,空速3200 h
‑1的条件下,转化率为20%, 选择性98%,充分显示了单原子pt催化剂对丙烷直接脱氢的优越性。
9.巩金龙课题组(acs catalysis, 2016, 6, 2158
‑
2162)报道了一种微量pt修饰的zno/al2o3催化剂,500 mg的催化剂在常压下,600℃的反应温度,空速为3 h
−1,c3h8/h
2 =1/1,氮气作为平衡气,总流量为50 ml/min。初始的平衡转化率为35%,4h后为31%,选择性94%,失活速率参数为15%。虽然pt作为助剂可以减少作为活性位点zn
2
的还原,从而达到不错的脱氢活性和最小化的pt使用量,但是积碳和不稳定的活性结构仍然导致明显的失活发生。
10.weckhuysen课题组(angewandte chemie
‑
international edition, 2014, 53,
ꢀ
9251
‑
9256)采用等体积浸渍法合成由1000 ppm pt,3 wt% ga,0.25 wt% k负载在al2o3的催化剂。常压下,0.15g的催化剂在620 o
c的反应温度,丙烷流速9 ml/min
‑1的条件下对催化剂进行了评价,首次循环中达到转化率42%,选择性97%,在每个循环15分钟后,需要在750 o
c下用6%的o2进行再生。掺杂的k由于覆盖了酸性位点,因此可以在一定程度上抑制积碳的产生,但通过在线的紫外可见拉曼光谱还是观察到产生了较多的积碳。并且pt再生前后的分散度没有发生明显变化,表明存在金属载体之间的很强相互作用。
11.taoufik(acs catalysis, 2018, 8, 7566
‑
7577)课题组采用接枝法将ga(i
‑
bu)3接枝到氧化铝和氧化硅载体上来研究不同活性位点的区别,指出al
‑
o
‑
ga活性位点比si
‑
o
‑
ga活性位点具有更好的异质裂解c
‑
h键的能力。但通过这种接枝的方式负载活性金属的催化剂表现出较差的活性和稳定性(初始转化率~24%,反应约25h后为~9%,选择性维持在约90%左右)。
12.陆安慧课题组(science, 2012, 335, 1205
‑
1208)报道通过原子层沉积的手段来沉积可控的al2o3涂层可以很好地调节pd/al2o3催化剂表面,并用于高温下乙烷的氧化脱氢反应性能研究。由于al2o3原子层的限制作用,pd粒子的抗烧结能力大大提高,同时产物分子对pd物种的吸附亲和力减弱,从而很好地提高了催化剂的抗烧结能力和抗积碳能力。
13.somorjai课题组(nature materials, 2009, 8, 126
‑
131)设计了一种稳定的耐高温的介孔无定形二氧化硅包裹pt纳米颗粒的核壳结构用于高温下co氧化和乙烯加氢研究,并显示出很好的稳定性。合成采用三步法的策略来实现:(1)以十四烷基三甲基溴化铵为封端剂,合成纳米pt颗粒;(2)二氧化硅在pt核周围聚合,生成pt@sio2介孔结构;(3)通过煅烧去除有机物分子,生成pt@sio2核壳型纳米粒子。
14.sch
ü
th课题组(angewandte chemie
‑
international edition, 2006, 118, 8404
‑
8407)通过两步法合成氧化锆包裹的au纳米颗粒,显示出很好的高温下催化性能。先合成au凝胶后还原得到均匀分布的au纳米颗粒,随后在au颗粒表面包裹二氧化硅,待硅壳生长完成后,获得单分散的au@sio2纳米球颗粒。在au@sio2纳米球表面涂抹表面活性剂后和正丁醇锆反应,从而包裹上氧化锆,煅烧后获得au@sio2@zro2。最后用naoh将sio2溶解,得到中空的au@ zro2催化剂。
15.以上研究表明,通过常规的合成策略来获得的pt基催化剂都存在反应温度高、转化率低、催化剂易失活等缺点。
技术实现要素:
16.本发明的目的在于提供一种丙烷直接脱氢制丙烯的催化剂及其制备方法,该催化剂在低温下具有高催化活性,高丙烯选择性,催化剂结构稳定,抗积碳能力强等优点。
17.本发明提供的催化剂各组分质量百分比分别为:1、pt:0.001~0.1%,ga:1%~10%,al:23.100%~51.635%,si:0.5~20%,o:44.200~49.475%。
18.本发明采用浸渍
‑
包覆法制备催化剂,其制备步骤如下:1)将商业γ
‑
al2o3在空气中以1~5 o
c/min的升温速率升至700 o
c,维持2~3h,在0.01mpa低压下,抽真空2h,备用。
19.2)称取一定质量的ga(no3)3·
xh2o,以pt/ga= 0.0001~0.1的质量比,称取pt(nh3)4(no3)2,溶于pt/h2o=0.000008~0.0008重量比的水中,记为a溶液。以pt/al2o3=0.00001~0.001的重量比称取al2o3。将a溶液逐滴加入到称取好的al2o3中,静置2~3 h,烘干,研磨,煅烧,制得ptga/al2o3催化剂。
20.3)以乙醇/氨水=20:1~40:1的体积比配置成b溶液,以ptga/al2o3(g)/氨水(ml)=0.15~0.3的比例称取ptga/al2o3加入到b溶液中,超声成悬浮液c。
21.4)以正硅酸乙酯(teos)(g)/乙醇(ml)=0.00058~0.02332的比例配置d溶液,以1~5ml/h的速率滴加入悬浮液c中,再搅拌10~20 h,离心得到产物。所得产物在353 k下烘干,研磨,在623~673 k下空气中煅烧2~4小时(升温速率1~2 o
c
·
min
‑1),得到质量百分比分别为:pt:0.001~0.1%,ga:1%~10%,al:23.100%~51.635%,si:0.5~20%,o:44.200~49.475%。
22.制得的催化剂的应用如下:将催化剂与石英沙以1:5的重量比混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1~2 h,目的是为了吹干净催化剂表面的杂质。以丙烷/氢气(体积比)=1:1,氩气作平衡气,组成的混合气为原料,反应温度为450 o
c,反应压力为0.1 mpa,空速1~3h
‑1下来合成丙烯。
23.发明原理:采用包裹无机氧化物的策略可以在物理空间上一定程度地阻碍金属颗粒的迁移,同时通过改变金属
‑
载体界面来改变ga2o3的还原能力,通过还原后不同价态的ga物种锚定高分散的pt位点,从而提高催化性能以及抗烧结和抗积碳能力。因此,通过使用超低负载量的pt作为活性相,基于氧化镓和氧化铝的强载体效应,以氧化镓为基底辐助形成高分散pt位点,在活性金属表面再包裹上二氧化硅壳层,从而制得高活性、高丙烯选择性、高稳定性且抗积碳的新型低温催化剂。
24.本发明提出的无定形二氧化硅在表层包裹的丙烷直接脱氢制丙烯的pt基催化剂,
与现有方法相比,其有益效果在于:1)催化剂活性组分颗粒尺寸小且均一分布,ga团簇均匀分散在al2o3载体上,高分散的pt活性位点牢固锚定在ga团簇上,同时在表面上被二氧化硅薄壳层限制迁移,形成结构稳定的pt活性位点结构,有利于提高低温下的催化活性,较高的丙烯选择性和催化剂的热稳定性。
25.2)通过无定形二氧化硅壳层表面包裹催化剂,有效地抑制裂解,聚合,深度脱氢等副反应的发生,减缓了催化剂的失活。
26.3)低负载量的高分散pt位点,有效提高了贵金属原子利用率的同时减低经济成本。
附图说明
27.图1 (a
‑
d)反应后的2.5%si@0.1pt5ga/al2o3催化剂的球差电镜图。
28.图2 (a
‑
c)反应后的0.1pt5ga/al2o3催化剂和(d
‑
f)反应后的2.5%si@0.1pt5ga/al2o3催化剂的球差电镜图。
29.图3 (a
‑
b)反应后的0.1pt5ga/al2o3催化剂和(c
‑
d)反应后的2.5%si@0.1pt5ga/al2o3催化剂的电镜图。
30.图4 (a
‑
f)反应后的2.5%si@0.1pt5ga/al2o3催化剂的球差电镜图。
31.图5 (a
‑
b)反应后的催化剂的drift
‑
co图。
具体实施方式
32.为了使本发明所述的内容更加便于理解,下面结合具体实施方式对本发明所述的技术方案做进一步的说明,但是本发明不仅限于此。
33.实施例1:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,36.68gga(no3)3·
xh2o 溶解于250 ml水中,制成pt、ga混合盐水溶液,记为a溶液。取2.5 ml溶液a,用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0.001pt5ga/al2o3。
34.称量0.3 g合成的0.001pt5ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.1166 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以3 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌15h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持4h,获得含量pt:0.001%;ga:5%;al:43.687%%;si:5%;o:46.312%的催化剂,记作5%si@0.001pt5ga/al2o3。将其造粒成40
‑
60目,备用。
35.在固定床中,将0.1 g(40
‑
60目)的5%si@0.001pt5ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)
条件下,将反应器床层温度升高至450 o
c并维持2h,然后通入丙烷:氢气=1:1(空速1h
‑1)进行催化评估。丙烷转化率5.8%,丙烯的选择性90.1%,失活速率常数0.059 h
‑1。
36.实施例2:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,3.668gga(no3)3·
xh2o 溶解于25 ml水中,制成pt、ga混合盐水溶液,记为a溶液。取2.5 ml溶液a,用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0.01pt5ga/al2o3。
37.称量0.3 g合成的0.01pt5ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.1166 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以3 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌15h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持4h,获得含量pt:0.01%;ga:5%;al:43.68%;si:5%;o:46.31%的催化剂,记作5%si@0.01pt5ga/al2o3。将其造粒成40
‑
60目,备用。
38.在固定床中,将0.1 g(40
‑
60目)的5%si@0.01pt5ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持2h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)进行催化评估。丙烷转化率9.5%,丙烯的选择性90.4%,失活速率常数0.043 h
‑1。
39.实施例3:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.3668gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 5ga/al2o3。
40.称量0.3 g合成的0.1pt5ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.0116 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以5 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌10h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持2h,获得含量pt:0.1%;ga:5%;al:48.7%;si:0.5%;o:45.7%的催化剂,记作0.5%si@0.1pt5ga/al2o3。将其造粒成40
‑
60目,备用。
41.在固定床中,将0.1 g(40
‑
60目)的0.5%si@0.1pt5ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持2h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)
进行催化评估。丙烷转化率22.5%,丙烯的选择性90.0%,失活速率常数0.019 h
‑1。
42.实施例4:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.3668gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt5ga/al2o3。
43.称量0.3 g合成的0.1pt5ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.0583 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以4 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌12h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持3h,获得含量pt:0.1%;ga:5%;al:46.4%;si:2.5%;o:46.0%%的催化剂,记作2.5%si@0.1pt5ga/al2o3。将其造粒成40
‑
60目,备用。
44.在固定床中,将0.1 g(40
‑
60目)的2.5%si@0.1pt5ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)进行催化评估。丙烷转化率23.0%,丙烯的选择性95.0%,失活速率常数0.008 h
‑1。
45.实施例5:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.3668gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 5ga/al2o3。
46.称量0.3 g合成的0.1pt5ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.1166 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以3 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌15h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持4h,获得含量pt:0.1%;ga:5%;al:43.6%;si:5%;o:46.3%的催化剂,记作5%si@0.1pt5ga/al2o3。将其造粒成40
‑
60目,备用。
47.在固定床中,将0.1 g(40
‑
60目)的5%si@0.1pt5ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持2h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)进行催化评估。丙烷转化率22.3%,丙烯的选择性93.0%,失活速率常数0.016 h
‑1。
48.实施例6:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.5868gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 8ga/al2o3。
49.称量0.3 g合成的0.1pt8ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,1 ml浓氨水,超声30 min,记作c溶液。再称量0.1166 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以3 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌15h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持4h,获得含量pt:0.1%;ga:8%;al:41.5%;si:5%;o:45.4%的催化剂,记作0.5%si@0.1pt8ga/al2o3。将其造粒成40
‑
60目,备用。
50.在固定床中,将0.1 g(40
‑
60目)的0.5%si@0.1pt8ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)进行催化评估。丙烷转化率22.8%,丙烯的选择性90.8%,失活速率常数0.019 h
‑1。
51.实施例7:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,7.33gga(no3)3·
xh2o 溶解于250 ml水中,制成pt、ga混合盐水溶液,记为a溶液。取2.5 ml溶液a,用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 001pt 1ga/al2o3。
52.称量0.3 g合成的0.001pt1ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,1 ml浓氨水,超声30 min,记作c溶液。再称量0.0116g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以5 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌10h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持2h,获得含量pt:0.001%;ga:1%;al:51.635%;si:0.5%;o:46.864%的催化剂,记作0.5%si@0.001pt1ga/al2o3。将其造粒成40
‑
60目,备用。
53.在固定床中,将0.1 g(40
‑
60目)的0.5%si@0.001pt1ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持2h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)进行催化评估。丙烷转化率12.3%,丙烯的选择性84.7%,失活速率常数0.087 h
‑1。
54.实施例8:
将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.3668gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 5ga/al2o3。
55.称量0.3 g合成的0.1pt5ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.2332 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以2 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌18h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以2 o
c/min的升温速率升至400 o
c,维持3h,获得含量pt:0.1%;ga:5%;al:37.9%;si:10%;o:47.0%的催化剂,记作10%si@0.1pt5ga/al2o3。将其造粒成40
‑
60目,备用。
56.在固定床中,将0.1 g(40
‑
60目)的10%si@0.1pt5ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)进行催化评估。丙烷转化率21.3%,丙烯的选择性91.6%,失活速率常数0.025 h
‑1。
57.实施例9:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.3668gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 5ga/al2o3。
58.称量0.3 g合成的0.1pt5ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.4664 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以1 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌20h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以2 o
c/min的升温速率升至400 o
c,维持4h,获得含量pt:0.1%;ga:5%;al:26.6%;si:20%;o:48.3%的催化剂,记作20%si@0.1pt5ga/al2o3。将其造粒成40
‑
60目,备用。
59.在固定床中,将0.1 g(40
‑
60目)的20%si@0.1pt5ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速1.2h
‑1)进行催化评估。丙烷转化率20.0%,丙烯的选择性89.4%,失活速率常数0.058 h
‑1。
60.实施例10:将商业γ
‑
al2o3在空气中以5 o
c/min的升温速率升至700 o
c,维持2h,真空烘箱
pt(nh3)4(no3)2,0.7336gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 10ga/al2o3。
67.称量0.3 g合成的0.1pt10ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,1 ml浓氨水,超声30 min,记作c溶液。再称量0.0583 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以4 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌12h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持3h,获得含量pt:0.1%;ga:10%;al:42.9%;si:2.5%;o:44.5%的催化剂,记作2.5%si@0.1pt10ga/al2o3。将其造粒成40
‑
60目,备用。
68.在固定床中,将0.1 g(40
‑
60目)的2.5%si@0.1pt10ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速3h
‑1)进行催化评估。丙烷转化率20.3%,丙烯的选择性93.8%,失活速率常数0.012 h
‑1。
69.实施例13:将商业γ
‑
al2o3在空气中以1 o
c/min的升温速率升至700 o
c,维持3h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.7336gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 10ga/al2o3。
70.称量0.3 g合成的0.1pt10ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.1166 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以3 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌15h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以1 o
c/min的升温速率升至350 o
c,维持4h,获得含量pt:0.1%;ga:10%;al:40.1%;si:5%;o:44.8%的催化剂,记作5%si@0.1pt10ga/al2o3。将其造粒成40
‑
60目,备用。
71.在固定床中,将0.1 g(40
‑
60目)的5%si@0.1pt10ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速3h
‑1)进行催化评估。丙烷转化率20.5%,丙烯的选择性91.3%,失活速率常数0.026 h
‑1。
72.实施例14:将商业γ
‑
al2o3在空气中以1 o
c/min的升温速率升至700 o
c,维持3h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.7336gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记
为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 10ga/al2o3。
73.称量0.3 g合成的0.1pt10ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.2332 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以2 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌18h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以2 o
c/min的升温速率升至400 o
c,维持3h,获得含量pt:0.1%;ga:10%;al:34.4%;si:10%;o:45.5%的催化剂,记作10%si@0.1pt10ga/al2o3。将其造粒成40
‑
60目,备用。
74.在固定床中,将0.1 g(40
‑
60目)的10%si@0.1pt10ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速3h
‑1)进行催化评估。丙烷转化率19.7%,丙烯的选择性88.9%,失活速率常数0.041 h
‑1。
75.实施例15:将商业γ
‑
al2o3在空气中以1 o
c/min的升温速率升至700 o
c,维持3h,真空烘箱0.01 mpa抽真空2h,备用。先称取2g煅烧后的γ
‑
al2o3置于50 ml烧杯中,再称取0.0040g的 pt(nh3)4(no3)2,0.7336gga(no3)3·
xh2o 溶解于2.5 ml水中,制成pt、ga混合盐水溶液,记为a溶液。用100 ul的移液枪将a溶液逐滴加入到烧杯中,边滴边用玻璃棒搅拌,待完全滴加完后,继续搅拌1h,用封口膜将烧杯密封,室温下静置3h,放入烘箱中80 o
c,12h。烘干后,研磨,在空气中以5 o
c/min的升温速率升至500 o
c,维持3h,获得的药品记作0. 1pt 10ga/al2o3。
76.称量0.3 g合成的0.1pt10ga/al2o3催化剂到100 ml圆底烧瓶中,加入40 ml无水乙醇,2 ml浓氨水,超声30 min,记作c溶液。再称量0.4664 g teos 溶解于20 ml无水乙醇中,记作d溶液。将d溶液吸入注射器中用微量注射泵以1 ml/min的滴加速率加入到含a溶液的圆底烧瓶中,此时的搅拌速度为1200转/分钟剧烈搅拌。滴完后继续搅拌20h。随后将溶液以4000转/分钟离心分离,放入烘箱80 o
c,12h。研磨后,在空气中以2 o
c/min的升温速率升至400 o
c,维持4h,获得含量pt:0.1%;ga:10%;al:23.1%;si:20%;o:46.8%的催化剂,记作20%si@0.1pt10ga/al2o3。将其造粒成40
‑
60目,备用。
77.在固定床中,将0.1 g(40
‑
60目)的20%si@0.1pt10ga/al2o3催化剂与0.5 g(40
‑
60目)石英砂均匀混合后,填装入内径为6 mm的固定床石英管中段。在纯氩气(20 ml
·
min
‑1)条件下,将反应器床层温度升高至450 o
c并维持1h,然后通入丙烷:氢气=1:1(空速3h
‑1)进行催化评估。丙烷转化率17.9%,丙烯的选择性79.8%,失活速率常数0.053 h
‑1。
78.图1 (a
‑
d)反应后的2.5%si@0.1pt5ga/al2o3催化剂的球差电镜图。图a球差电镜暗场图说明ga金属团簇在催化剂表面均匀分散;图b
‑
c根据原子序数衬度机理,团簇中的孤立的亮点为pt活性位点,亮点周围较暗的为ga物种;图d为球差电镜明场图,根据原子序数衬度机理,团簇中的孤立的暗点为pt活性位点,亮点周围较暗的为ga物种。
79.图2 (a
‑
c)反应后的0.1pt5ga/al2o3催化剂和 (d
‑
f) 反应后的2.5%si@0.1pt5ga/
al2o3催化剂的球差电镜图。通过图(a
‑
c)说明反应后的0.1pt5ga/al2o3催化剂的ptga团簇发生明显团聚,而图(d
‑
f)说明反应后的2.5%si@0.1pt5ga/al2o3催化剂保持了高度分散的ptga团簇。对比说明高分散的pt位点被ga锚定,同时被二氧化硅壳层能有效地限制迁移。
80.图3 (a
‑
b)反应后的0.1pt5ga/al2o3催化剂和(c
‑
d)反应后的2.5%si@0.1pt5ga/al2o3催化剂的电镜图,(a
‑
b)虚线微al2o3边界,(c
‑
d)虚线为外延生长的二氧化硅壳层。说明二氧化硅壳层生长在了0.1pt5ga/al2o3催化剂的表面。
81.图4 (a
‑
f)反应后的2.5%si@0.1pt5ga/al2o3催化剂的球差电镜图,说明si, ga, al和pt元素在表面分散得很均匀,并且二氧化硅壳层紧密地包裹在ptga/ al2o3催化剂表面。
82.图5 (a
‑
b)反应后的催化剂的drift
‑
co图,说明pt位点在反应后还是以高分散的pt位点形式存在,并且由于二氧化硅壳层和pt位点的相互作用导致峰有所偏移,说明调控了pt位点的电子性质。
83.以上五图的催化剂均为反应后的催化剂,说明了高分散的pt位点锚定在高分散的ga团簇上和二氧化硅壳层紧密地包裹在ptga/al2o3表面,同时也说明了二氧化硅壳层可以很好地限制活性位点在反应过程中的迁移,从而起到抵制烧结的作用。
84.以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。