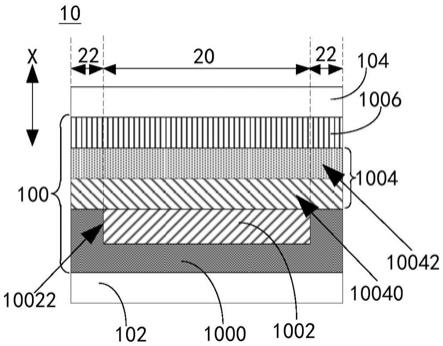
多层胶带
1.本发明涉及广泛地用于接合结构部件和设备组件的胶带的技术领域。本发明更特别地涉及具有特殊交联的压敏胶粘剂为特征的发泡的胶带。
2.本发明尤其针对与粘附技术相关的参数“润湿(润湿性)”。下面润湿理解为是指形成在压敏胶粘剂和待粘合基材之间的界面。因此,术语“润湿”描述了压敏胶粘剂补偿表面的不平整性和/或曲率以及在其自身和基材之间置换(排挤)空气的能力。润湿越强,就能够越有效地形成在压敏胶粘剂和基材之间的相互作用并且粘附性和胶粘性越好。尤其是对粗糙表面或具有与生产有关的不平整性或曲率或波纹的表面,经常观察到,作为机械载荷的结果,曾经实现的润湿再次变弱,换言之,发生去润湿。保持发生弹性变形以顺应于粘合区域的表面轮廓(其可为弯曲的、粗糙的等等)的柔性制品(典型地以膜、塑料或金属板/或泡沫膜/板的形式)在恢复至其原始形状时抵抗施加的排斥力能力(即,承受制品的排斥力的能力),经常也被称为“抗排斥性”。
3.此外,润湿应不同于随着时间的推移形成粘合力(剥离粘附性)。即使当初始润湿良好时,粘合力也可随时间进一步提高,因为越来越多的能够与表面相互作用的官能团朝着表面定向。
4.对于不同应用领域,例如在建筑业中、在技术产品的工业制造中、或用于组装目的,需要越来越厚但也强力粘合的胶带(称为“组装胶带”)。由于粘合经常在户外进行和/或粘合产品暴露在外部天气条件下,因此对这样的胶带的性质的预期经常是高的。因此,粘合应为强力的、持久的和耐候性的;在许多情况下,需要高的耐湿性、耐热性以及对组合的热和湿的耐受性。而且,胶带应迅速润湿,并且在这种情况下,补偿粘合接合部中和/或待粘合基材上的不平整性,并且胶带应从开始呈现出高的粘合力(初始粘合力)。当使用未发泡胶带时,有效润湿的另一优点是它允许粘合透明材料而没有光学缺陷,如即使对于厚的胶带也日益期望的(例如,在透明材料例如玻璃或透明塑料的粘合的领域中)。
5.用于这样的目的胶带通常配备有这样的胶粘剂,对于其而言技术胶粘剂性质必须非常好地彼此匹配。例如,必须非常精细地调整内聚性、初始粘性、流动行为、和其它性质。考由于影响这些性质的压敏胶粘剂的技术形式经常对各性质具有不同的影响,因而精细的调整通常是困难的,或者在结果中必须接受折衷。
6.此外,特别是对于非常厚的胶带,实现高度均匀的产品经常是困难的;作为加工的结果,非常厚的胶带经常在整个层中不均匀。这通常是不希望的,因为经常需要具有明确定义的性质的胶带而不管其层厚和其制造如何。
7.具有适合用于压敏胶粘剂应用的粘弹性性质的物质在如下方面是引人注意的:它们在机械变形时既产生粘性流动又产生弹性恢复力。这两个过程就其各自的比例而言彼此成一定的关系,这不仅取决于所讨论的物质的精确组成、结构和交联度,而且还取决于形变的速率和持续时间、以及温度。
8.成比例(按一定比例)的粘性流动对于粘附性的实现是必要的。仅由具有相对高的迁移性的大分子产生的粘性分量(组分)允许有效地润湿待粘合基材并有效地流动到待粘合基材上。高粘性流动分量导致高的固有胶粘性(也称为压敏胶粘性或粘性),并因此也常
常导致高的粘合力。高度交联的体系、结晶的聚合物或已经历玻璃状固化的聚合物由于缺乏可流动分量而通常缺乏固有胶粘性。
9.成比例(按一定比例)的弹性恢复力对于内聚性的实现是必要的。它们例如由链非常长的且高度缠结的大分子以及由物理或化学交联的大分子产生,并且它们允许传递作用在胶粘粘合上的力。它们导致胶粘粘合在足够的程度上承受作用在其上的持续载荷(例如以长期的剪切载荷的形式)相对长的时间段。
10.在发泡的多层胶带中,持续载荷可导致应力的不均匀分布,如果该力大于压敏胶粘剂层对表面的粘附力,则其表现为压敏胶粘剂层的部分分离。因此,被润湿的区域的比例变得更小。
11.为了防止压敏胶粘剂从基材流掉(流出),并且为了保证粘合组件中的压敏胶粘剂的足够的稳定性,足够的压敏胶粘剂的内聚性因此是必要的。然而,对于良好的粘附性质,压敏胶粘剂另外必须能够流到基材上,在边界层中与表面充分地形成相互作用,并且保证基材表面的有效且持久的润湿。而且,为了防止粘合接合部内(在压敏胶粘剂层内)的破裂,就压敏胶粘剂而言需要一定的弹性。
12.为了实现压敏胶粘剂的足够的内聚性,通常使它们交联,即,单独的大分子通过桥接键合而彼此连接。交联可以各种方式完成:例如,存在物理和化学(热)交联方法。
13.化学交联方法通常导致不可逆的经常为共价的网络,这确保了足够的内聚性,特别是在高温下。然而,由于该网络的不可逆转性,应力例如由机械形变引起的应力无法降低或消散,且作为聚合物链的减少的迁移性的结果,粘附性下降。这可导致胶粘剂在持续载荷下的分离(去润湿)。
14.为了产生均匀的胶带,使聚合物经受热交联是有利的:即使对于厚的层,也可容易地均匀地供应热能。相比之下,已经通过光化辐射(例如紫外线或电子束)交联的组合物层在整个交联的层中呈现出交联分布(曲线)。该交联分布由如下的事实产生:辐射在其穿透到该层中的深度方面是有限的,其中由于吸收过程,辐射的强度也随着穿透的深度而降低。因此,辐射化学交联的组合物层的外部区域与位于更内部的区域相比更强地交联,其中交联强度整体上朝向内部减小。特别地对于厚的层,该效果是非常显著的。
15.例如,ep 2 305 389 a2和ep 2 617 789 a1描述了具有良好的胶粘性和内聚性性质的、热交联的、发泡和未发泡的组装胶带。然而,这些胶带在其润湿行为方面以及在对弯曲基材的粘合方面呈现出弱点,特别是如果所述基材具有低的表面能的话。
16.wo 2013/048985 a2和wo 2013/048945 a1描述了多层组装胶带,其特别适合用于对非极性表面例如汽车饰面的粘合。wo 2013/048985 a2的胶带的特征在于,外部psa(压敏胶粘剂)层包括(甲基)丙烯酸酯,其具有拥有12至32个碳原子的2
‑
烷基链烷醇残基(基团),以及任选地具有c1
‑
12链烷醇残基。在wo 2013/048945 a1中,外部psa层特别地包括具有伯醇残基的丙烯酸酯,所述伯醇残基具有14至25个碳原子和至少2至最多4的iso指数。除了其中描述的产品使用uv辐射交联的缺点之外,还发现,初始良好的润湿在载荷下劣化,且因此发生去润湿。
17.us 2011/0244230 a1描述了基于丙烯酸酯的泡沫胶带,其特别柔软且高度适合用于对不平整基材的粘合。然而,所述胶带通过uv辐射交联,从而产生的交联梯度导致相对差的润湿行为。
18.ep 2 226 372 a1描述了热交联的psa,其包括具有8
‑
15重量%的丙烯酸浓度的聚丙烯酸酯且其特征在于,线性丙烯酸酯与支化丙烯酸酯的比率在1:6至10:1质量分数的范围内。在根据该发明的所有实施例中,使用配位热交联剂例如乙酰丙酮铝(iii),其导致可逆网络。然而,该组合物对于用作组装胶带显示出不足的温度稳定性。
19.本发明的目的是提出这样的强力胶带,其迅速润湿具有不同表面能的粗糙和/或弯曲的表面例如金属、塑料如abs或聚碳酸酯、和汽车饰面,并且在此形成高粘附性。此外,用其产生的粘合具有良好的剪切强度(即使在升高的温度下)、高的对热和湿的耐受性、以及在动态载荷下的高粘合强度,后者特别是在低温下。最后,粘合上的持续的机械载荷将不应导致胶带从表面去润湿。
20.该目的的实现基于如下的构思:发泡载体设置有双重交联的聚(甲基)丙烯酸酯psa。因此,本发明的第一和一般主题是胶带,其包括:
21.至少一个发泡层和
22.至少一个压敏胶粘剂层,其中所述压敏胶粘剂层包括至少一种聚(甲基)丙烯酸酯并且所述聚(甲基)丙烯酸酯是用至少一种共价交联剂和至少一种配位交联剂交联的。
23.本发明的胶带的特征特别地在于,迅速润湿和对低能表面的高的去润湿耐受性(抗排斥性)(即使在粘合上的持续机械载荷下也是如此),以及在其它方面良好的技术胶粘剂性质。
24.发泡层优选地在达到至少50重量%、更优选地至少70重量%、非常优选地至少80重量%、更特别地至少90重量%的程度上包括至少一种聚合物,在各自的情况下基于发泡层的总重量,所述聚合物选自橡胶、更特别地天然橡胶,聚氨酯,聚(甲基)丙烯酸酯和苯乙烯嵌段共聚物,以及所述聚合物的共混物。更优选地,发泡层在达到至少50重量%、更优选地至少70重量%、非常优选地至少80重量%、更特别地至少90重量%的程度上包括一种或多种聚(甲基)丙烯酸酯,在各自的情况下基于发泡层的总重量。
[0025]“聚(甲基)丙烯酸酯”是可通过丙烯酸类和/或甲基丙烯酸类单体、以及任选地另外的可共聚单体的自由基聚合获得的聚合物。更特别地,“聚(甲基)丙烯酸酯”是这样的聚合物,其单体基础在达到至少50重量%的程度上由丙烯酸、甲基丙烯酸、丙烯酸酯和/或甲基丙烯酸酯组成,其中丙烯酸酯和/或甲基丙烯酸酯至少按比例地、优选地在达到至少30重量%的程度上存在,基于所讨论的聚合物的总的单体基础。
[0026]
更特别地,发泡层在达到至少50重量%、更优选地至少70重量%、非常优选地至少80重量%、更特别地至少90重量%的程度上包括至少一种聚(甲基)丙烯酸酯a,在各自的情况下基于发泡层的总重量,聚(甲基)丙烯酸酯a可得自以下单体组成:
[0027]
a1)40
‑
77重量%的至少一种具有不超过
‑
40℃的均聚物玻璃化转变温度的(甲基)丙烯酸酯,其醇组分基于支化的伯醇;
[0028]
a2)20
‑
40重量%的至少一种其醇组分基于线性c1‑
c
18
醇的(甲基)丙烯酸酯;
[0029]
a3)0
‑
20重量%的至少一种其醇组分基于环状醇的(甲基)丙烯酸酯;
[0030]
a4)3
‑
20重量%的丙烯酸。
[0031]
存在于发泡层中的聚合物、特别地聚合物a优选具有至少500 000g/mol、更优选地至少700 000g/mol的重均分子量mw。同样优选地,存在于发泡层中的聚合物具有不超过1 700 000g/mol的重均分子量mw。对于存在于发泡层中的聚合物,多分散性pd,即作为重均分
子量mw和数均分子量mn的商确定的摩尔质量分布的幅度,优选为10≤pd≤100、更优选地20≤pd≤80。
[0032]
发泡层优选地为热交联的,导致该层的非常均匀的形成。特别优选地,发泡层通过至少一种缩水甘油醚、更特别地至少一种3
‑
缩水甘油氧基丙基三烷氧基硅烷和/或多缩水甘油醚热交联,非常优选地至少通过3
‑
缩水甘油氧基丙基三乙氧基硅烷和/或季戊四醇四缩水甘油醚热交联。优选地与作为促进剂的胺、更优选地与作为促进剂的3
‑
氨基丙基三乙氧基硅烷和/或异佛尔酮二胺组合,进行发泡层的交联。
[0033]
待交联的发泡层中的全部交联剂的分数优选为最高达1重量%、更优选地最高达0.8重量%、更特别地0.05
‑
0.6重量%、和非常优选地0.1
‑
0.5重量%,在各自的情况下基于待交联的聚合物的总量。
[0034]
待交联的发泡层中的全部促进剂的分数优选为0.1
‑
1.5重量%、更优选地0.15
‑
1.2重量%,在各自的情况下基于待交联的聚合物的总量。
[0035]
发泡层中的胺促进剂的存在并不重要,尤其是在三层或多层构造的情况下,因为在这些构造中,发泡层充当载体层且因此通过位于外部的胶粘剂/psa层在很大程度上屏蔽了氧化物质如大气氧的影响。
[0036]
发泡层原则上可以任何期望的方式和方法发泡。例如,发泡层可已借助于引入其中或在其中释放的推进剂气体发泡。引入的推进剂气体在此包括例如co2或n2,任选地也为超临界流体的形式。
[0037]
替代地,可借助于在气体释放时热分解的发泡剂来实现推进剂气体的释放,实例为nahco3,柠檬酸、抗坏血酸、富马酸、葡萄糖酸或乳酸的游离酸或衍生物,或放热的发泡剂如氮杂胆管胺。
[0038]
还合适的是机械发泡(起泡)。
[0039]
发泡层优选地包括至少一种选自如下的发泡剂:空心聚合物球、实心聚合物球、空心玻璃球、实心玻璃球、空心陶瓷球、实心陶瓷球和实心碳球。更优选地,发泡层包括至少部分膨胀的空心微球。这些为至少部分膨胀的微球,所述微球在其基本状态下是弹性和可膨胀的并且具有热塑性聚合物壳。这些球在基本状态下填充有低沸点液体或液化气体。所用壳材料特别地为聚丙烯腈、pvdc、pvc或聚丙烯酸酯。常见的低沸点液体特别地为低级烷烃的烃,例如异丁烷或异戊烷,其在压力下以液化气体的形式被包封在聚合物壳中。对于这种的微球,术语“微气球”也是惯常的。
[0040]
使微气球暴露于热导致聚合物外壳软化。同时,壳内的液体形式的推进剂气体经历向其气态的转变。当这发生时,微气球不可逆地拉伸并经历三维膨胀。当内部和外部压力相互匹配时,膨胀结束。由于保留了聚合物壳,结果是闭孔泡沫体。
[0041]
多种类型的微气球是可商购的,且主要在其尺寸(在未膨胀状态下直径6
‑
45μm)方面和在它们膨胀所需的起始温度(75
‑
220℃)方面不同。未膨胀的微气球类型也可以具有约40
‑
45重量%的固体分数或微气球分数的水分散体的形式获得,且另外还可以聚合物结合的微气球(母料)的形式获得,例如以约65重量%的微气球浓度在乙烯
‑
乙酸乙烯酯中。与未膨胀的微气球一样,微气球分散体和母料二者都适合这样用于生产发泡层。
[0042]
发泡层也可使用所谓的预膨胀空心微球来生产。对于该群组,在引入聚合物基体之前进行膨胀。
[0043]
术语“至少部分膨胀的空心微球”在本发明中被理解为意指,空心微球已经经历至少这样程度的膨胀,使得与包含未膨胀空心微球的相同的层相比在技术上有意义的程度上产生所讨论的层的密度的降低。这意味着微气球无需一定经历完全膨胀。“至少部分膨胀的空心微球”在各自的情况下优选地膨胀至其在未膨胀状态下的最大范围(尺寸)的至少两倍。
[0044]
表述“至少部分膨胀”涉及单独的空心微球的膨胀状态且不意图意指仅一部分所讨论的空心微球必须经历(初始)膨胀。因此,如果在发泡层中存在“至少部分膨胀的空心微球”和未膨胀的空心微球,这意味着未膨胀(完全未膨胀,换句话说甚至尚未经历初始膨胀)的空心微球不属于“至少部分膨胀的空心微球”。
[0045]
发泡层可包括二氧化硅,优选地用二甲基二氯硅烷表面改性的沉淀二氧化硅。这是有利的,因为由此允许调整该层的热剪切强度,更特别地增加该层的热剪切强度。此外,二氧化硅可出色地用于透明层。二氧化硅优选地以最高达15重量%存在于发泡层中,基于发泡层中存在的所有聚合物的全部。
[0046]
发泡层还可包括至少一种增塑剂。增塑剂优选地选自(甲基)丙烯酸酯低聚物、邻苯二甲酸酯、环己烷二羧酸酯(例如,dinch,来自basf)、水溶性增塑剂、增塑树脂、磷酸盐(酯)(例如,dmpp,来自lanxess)和多磷酸盐(酯)。
[0047]
如在一般使用中惯常的,压敏胶粘剂在本发明中被理解为这样的材料,其特别地在室温下是永久粘性的以及胶粘性的。压敏胶粘剂的特征是,可将它通过压力施加到基材并在那里保持粘附,而无需进一步限定要施加的压力或暴露于该压力的时间。在一些情况下,取决于压敏胶粘剂的精确性质以及基材、温度和大气湿度,短时间的最小压力的影响(其不超出短暂的轻柔接触)足以达到粘附效果,而在其他情况下,长时间暴露于高压也可能是必要的。
[0048]
压敏胶粘剂具有特定的特征性的粘弹性性质,其导致永久粘性和胶粘性。这些胶粘剂的特征是,当它们发生机械形变时,存在粘性流动过程并且还存在弹性恢复力的形成。这两个过程就其各自的比例而言彼此具有一定的关系,这不仅取决于压敏胶粘剂的精确组成、结构和交联度,而且还取决于形变的速率和持续时间、以及温度。
[0049]
成比例(按一定比例)的粘性流动对于粘附性的实现是必要的。仅由具有相对高迁移性的大分子产生的粘性分量(组分)允许有效地润湿将进行粘合的基材并有效地流动到将进行粘合的基材上。高粘性流动分量导致高的压敏胶粘性(也称为粘性或表面粘着性),并因此常常也导致高的粘附性。高度交联的体系、结晶的聚合物或具有玻璃状固化的聚合物缺乏可流动分量且通常缺乏粘性或至少仅具有很小的粘性。
[0050]
成比例(按一定比例)的弹性恢复力对于内聚性的实现是必要的。它们例如由非常长链的具有高缠结度的大分子以及由物理或化学交联、特别地不可逆交联的大分子产生,并且它们允许传递作用在胶粘粘合上的力。作为这些恢复力的结果,胶粘粘合能够在足够的程度上在相对长的时间段承受作用在其上的长期载荷,例如以持续的剪切载荷的形式。
[0051]
为了更精确地描述和量化弹性分量和粘性分量的程度、以及各分量之间的关系,使用储能模量(g’)和损耗模量(g”)的变量,并且可借助于动态机械分析(dma)测定。g’是物质的弹性分量的量度,g”是物质的粘性分量的量度。两个变量都取决于形变频率和温度。
[0052]
所述变量可使用流变仪测定。在该情况下,例如将待研究的材料在板/板装置中暴
露于正弦振荡的剪切应力。在使用剪切应力控制操作的仪器的情况下,作为时间的函数测量形变,和相对于剪切应力的引入测量该形变的时间偏移。该时间偏移被称为相位角δ。
[0053]
储能模量g’如下定义:g'=(τ/γ)
·
cos(δ)(τ=剪切应力,γ=形变,δ=相位角=剪切应力向量和形变向量之间的相位移)。损耗模量g”的定义如下:g”=(τ/γ)
·
sin(δ)(τ=剪切应力,γ=形变,δ=相位角=剪切应力向量和形变向量之间的相位移)。
[0054]
当在室温下(在该情况下根据定义在23℃下),在100‑
101rad/秒(弧度/秒)的形变频率范围内g’和g”两者都至少部分地位于103‑
107pa的范围内时,物质通常被认为是压敏胶粘性的,并且就本发明而言如此定义。“部分地”意味着,g’曲线的至少一段(部分)位于由100(包括端点)直至101(包括端点)rad/秒的形变频率范围(横坐标)以及103(包括端点)至107pa(包括端点)的g’值范围(纵坐标)所跨越的窗口之内,并且g”曲线的至少一段同样位于相应的窗口内。
[0055]
本发明的胶带的psa层优选地包括至少一种聚(甲基)丙烯酸酯b,所述聚(甲基)丙烯酸酯b可得自以下单体组成:
[0056]
b1)55
‑
75重量%的至少一种具有不超过
‑
40℃的均聚物玻璃化转变温度的(甲基)丙烯酸酯,其醇组分基于支化的伯醇;
[0057]
b2)20
‑
40重量%的至少一种其醇组分基于线性c1‑
c
18
醇的(甲基)丙烯酸酯;
[0058]
b3)5
‑
15重量%的丙烯酸。
[0059]
psa层的聚(甲基)丙烯酸酯优选地具有至少500 000g/mol、更优选地至少700 000g/mol的重均分子量mw。同样优选地,psa层的聚(甲基)丙烯酸酯具有不超过1 700 000g/mol的重均分子量mw。对于psa层的聚(甲基)丙烯酸酯,多分散性pd,即作为重均分子量mw与数均分子量mn的商确定的摩尔质量分布的幅度,优选为10≤pd≤100、更优选地20≤pd≤80。
[0060]
聚合物的交联特别地涉及这样的反应,其中许多初始线性或支化的大分子通过在单独的大分子之间形成桥而连接,以形成或多或少支化的网络。桥接在此特别地通过如下方式实现:合适的化学分子(称为交联剂或交联剂物质)与大分子、例如与大分子的特别适合于被相应的交联剂分子攻击的某些官能团反应。攻击大分子的交联剂分子的位点通常称为“反应中心”。交联剂分子可将两个大分子彼此连接,方式是,一个且相同的(同一个)交联剂分子与两个不同的大分子反应,即特别地具有两个反应中心;然而,它们可具有超过两个反应中心,使得单个交联剂分子也可将三个或更多个大分子彼此连接。当同一个交联剂分子以其反应中心的至少两个攻击同一个大分子时,副反应可包括分子内反应。就聚合物的有效交联而言,这样的副反应通常是不希望的。
[0061]
本发明的胶带的psa层的聚(甲基)丙烯酸酯是用两种不同种类的交联剂交联的,即,
[0062]
1)用至少一种共价交联剂,这些为这样的交联剂,其共价地攻击待连接的大分子且因此在其相应的反应中心和大分子上的攻击位点(特别地官能团)之间形成共价化学键。原则上适用于此目的是形成共价键的所有可想到的化学反应。
[0063]
2)用至少一种配位交联剂,这些为这样的交联剂,其配位地攻击待连接的大分子且因此在其相应的反应中心和大分子上的攻击位点(特别地官能团)之间形成配位键。原则上适用于此目的是形成配位键的所有可想到的化学反应。
[0064]
本发明的胶带的psa层的共价交联剂在各自的情况下优选地选自多官能缩水甘油胺,环氧化物,特别地包括环氧化物官能化的有机硅烷,氮丙啶和异氰酸酯。
[0065]
优选的多官能缩水甘油胺为n,n,n',n'
‑
四(2,3
‑
环氧基丙基)环己烷
‑
1,3
‑
二甲基胺(例如,syna epoxy s610,synasia)和n,n,n',n'
‑
四(2,3
‑
环氧基丙基)
‑
间
‑
二甲苯
‑
α,α'
‑
二胺(例如,erisys ga
‑
240,cvc)。
[0066]
优选的多官能环氧化物为环氧基环己基羧酸2,2
‑
双(羟甲基)
‑
1,3
‑
丙二醇酯和3,4
‑
环氧基环己基羧酸(3,4
‑
环氧基环己烷)甲酯,以及环氧官能化的有机硅烷,特别地(3
‑
缩水甘油氧基丙基)三甲氧基硅烷(glymo)和(3
‑
缩水甘油氧基丙基)三乙氧基硅烷(glyeo)。
[0067]
优选的多官能氮丙啶是三羟甲基丙烷三(2
‑
甲基
‑1‑
氮丙啶丙酸酯)。
[0068]
优选的异氰酸酯是甲苯二异氰酸酯(tdi)、2,4
‑
甲苯二异氰酸酯二聚体、亚萘基1,5
‑
二异氰酸酯(ndi)、邻甲苯二异氰酸酯(todi)、二苯基甲烷二异氰酸酯(mdi)、三苯基甲烷三异氰酸酯、三(对异氰酸苯酯)硫代亚磷酸盐、和聚亚甲基聚苯基异氰酸酯。
[0069]
更优选地,共价交联剂包括至少一种包含至少一个环醚官能团、更特别地至少两个环醚官能团的化合物。
[0070]
本发明使用至少一种共价交联剂,然而两种或更多种共价交联剂的使用也是可能的,确切地不仅来自一种化合物类别的两种或更多种交联剂,而且还有来自不同化合物类别的共价交联剂的组合。
[0071]
本发明的胶带的psa层的配位交联剂优选地选自螯合物化合物、更特别地选自多价金属螯合物化合物。术语“多价金属螯合物化合物”是指其中多价金属与一种或多种有机化合物配位键合的那些化合物。更优选地,配位交联剂是其多价金属离子选自如下的多价金属螯合物化合物:al(iii)、zr(iv)、co(ii)、cu(i)、cu(ii)、fe(ii)、fe(iii)、ni(ii)、v(ii)、v(iii)、v(iv)、v(v)、zn(ii)、in(iii)、ca(ii)、mg(ii)、mn(ii)、y(iii)、ce(ii)、ce(iv)、st(ii)、ba(ii)、mo(ii)、mo(iv)、mo(vi)、la(iii)、sn(ii)sn(iv)和ti(iv),更特别地选自al(iii)、zr(iv)和ti(iv)。
[0072]
配位交联剂的配体原则上可为所有已知的配体。然而,用于有机化合物的配位键合的原子更特别地为具有自由电子对的那些原子,例如氧原子、硫原子、氮原子等。配位交联剂的有机化合物优选地选自烷基酯、醇、羧酸、醚和酮。更优选地,本发明的胶带的psa层的配位交联剂选自二丙氧基双(乙酰丙酮)钛、二丁氧基双(乙醇酸亚辛酯)钛(二丁氧基双(辛二醇乙醇酸酯)钛,titandibutoxidbis(octylenglycolat))、二丙氧基双(乙酰乙酸乙酯)钛、二丙氧基双(乳酸)钛、二丙氧基双(三乙醇胺)钛、二正丁氧基双(三乙醇胺)钛、三正丁氧基单硬脂酸钛、钛酸丁酯二聚体、聚(乙酰丙酮钛)、二异丙氧基单乙酸乙酯铝、二正丁氧基单乙酰乙酸甲酯铝、二异丁氧基单乙酰乙酸甲酯铝、二正丁氧基单乙酰乙酸乙酯铝、二仲丁氧基单乙酰乙酸乙酯铝、三乙酰丙酮铝、单乙酰丙酮双(乙酰丙酮酸乙酯)铝、和四乙酰丙酮锆;更特别地选自三乙酰丙酮铝和二异丙氧基单乙酸乙酯铝。
[0073]
在本发明中,使用至少一种配位交联剂;然而,也可使用两种或更多种配位交联剂,确切地不仅来自一种化合物类别的两种或更多种交联剂,而且还有来自不同化合物类别的配位交联剂的组合。
[0074]
优选地,在交联的开始之前,共价交联剂和配位交联剂以这样的比例存在于psa层中,使得基于配位交联剂的结合位点,共价交联剂的官能团摩尔过量地存在。更优选地,在
此共价交联剂的官能团对配位交联剂的结合位点的摩尔比,即,共价交联剂的官能团的所使用的物质的量n
共价
对配位交联剂的结合位点的所使用的物质的量n
配位
的比率,为3:1至9:1,相应地3≤n
共价
/n
配位
≤9,更特别地4.5:1至8.5:1。
[0075]
本发明的胶带的psa层可包括一种或多种增塑剂。增塑剂优选地选自(甲基)丙烯酸酯低聚物、邻苯二甲酸酯、环己烷二羧酸酯(例如,dinch,来自basf)、水溶性增塑剂、增塑树脂、磷酸盐(酯)(例如,dmpp,来自lanxess)、和多磷酸盐(酯)。
[0076]
在本发明的胶带的结构中,优选地在发泡层的两侧上布置psa层,其中psa层的至少一个已经根据本发明交联。
[0077]
特别优选地,根据本发明交联的psa层设置在发泡层的两侧上。这是有利的,因为在该情况下,胶带的两侧具有根据本发明的交联的psa层的非常良好的技术胶粘剂性质。在该情况下,更特别地,这两个psa层包括相同浓度的相同添加物,例如,功能性添加物和/或填料。两个psa层也可不含功能性添加物和/或填料。
[0078]
在一种实施方式中,在发泡层的两侧上布置根据本发明交联的psa层,并且所述psa层在化学上、在物理上和/或在它们的尺寸上相同。更特别地,两个psa层完全相同,其中忽略由与生产有关的不精确性和由其它类似的来源导致的那种微不足道的差异,例如可能由在普遍浓度范围内的杂质。
[0079]
不仅发泡层(条件是其表面之一暴露),而且一个或两个psa层均可用剥离衬垫或以其它方式用常规膜材料进行稳定化和/或保护。然而,剥离衬垫或以其它方式的膜材料不被认为是本发明的胶带的一部分,仅被认为是用于这样的带的储存、运输等的辅助物。
[0080]
在其中发泡层的一个表面暴露的本发明的胶带的实施方式中,在足够厚度的发泡层的情况下,发泡层的远离psa层且因此暴露的这一侧可通过如下被稳定化:使用具有低穿透深度的交联过程进行强交联,使得仅部分发泡层强交联,而在泡沫体的另一侧(面向psa层的侧)上,保留了原始存在的性质、更特别地粘弹性性质。
[0081]
不仅包含在本发明的胶带的psa层中的聚(甲基)丙烯酸酯、而且包含在本发明的胶带的发泡层中的那些可优选地通过自由基聚合、更特别地在溶液中根据现有技术制备。在任何由熔体后续加工的情况下,在聚合后去除溶剂。
[0082]
优选地将发泡层由熔体成形为层。在该情况下,优选地,存在发泡层的热交联。也可将psa层由熔体成形。然而,由于这些层典型地仅以最高达约100μm的厚度生产,它们也可出色地从溶液涂覆并在此后进行干燥。
[0083]
出于技术工艺的原因,与由聚合物溶液相比,可由熔体(在所谓的热熔体工艺中)远远更有效地生产非常厚的聚合物层,例如本发明的胶带的发泡层。关于无定形聚合物(例如聚丙烯酸酯)的熔体的定义,本发明使用f.r.schwarzl,polymermechanik:struktur und mechanisches verhalten von polymeren[polymer mechanics:structure and mechanical behavior of polymers],springer verlag,berlin,1990中规定的标准,根据该标准,粘度具有至多η≈104pa.s的数量级且内阻尼达到≥1的tanδ值。
[0084]
如果本发明的胶带的发泡层以及(在适当的情况下)psa层由熔体通过涂覆来生产,则产生由优选的热交联导致的问题。一方面,为了引发随后的热交联,必须在涂覆之前
添加热交联剂;另一方面,在该情况下,交联剂则暴露于用于产生和保持聚合物熔体的高温。甚至在受控交联的开始之前,这可导致聚合物的不受控的交联(称为胶凝)。为了尽可能抑制这种胶凝,热熔体工艺典型地使用非常缓慢反应的交联剂,并且仅在涂覆之前不久使用它们。然而,为了在涂覆后达到令人满意的交联结果,此外经常掺混所谓的促进剂。
[0085]
对于从溶液涂覆且将进行热交联的聚合物体系,促进剂的使用也可为意义的并且经常实施。热引发的交联程序通常与从施加的层热除去溶剂(即组合物层的干燥)相关。例如,作为起泡的结果,过度迅速地除去溶剂在此导致不良形成的、不平整且不均匀的层。出于该原因,干燥优选地在温和温度下进行。然而,为了保证良好的且足够快进行的交联,通常也将促进剂添加至溶剂体系。
[0086]
当所得层的厚度不是很大时,从溶液涂覆经常是优选的,从而存在与如下相关的显著问题:与聚合物溶液相比,待施加的聚合物熔体的粘度增加。
[0087]
在本发明中,发泡层优选地在促进剂的参与下交联。作为促进剂或具有促进效果的物质,特别地使用质子受体、电子对供体(路易斯碱)和/或电子对受体(路易斯酸)。促进剂是通过对应于技术目标的足够的反应速率来支持交联反应的化合物或化学品。这特别地以催化的方式(通过交联反应的活化)和/或通过如下方式实现:将交联剂物质中或待交联大分子中的官能团转化为能够在大分子的连接反应的意义上彼此反应(桥接,网络形成)或经由交联剂物质与其它官能团反应的官能团。
[0088]
促进剂本身不参与这种连接反应,即它们本身不交联,但能够以反应产物或片段的形式引入到网络中或连接至其。因此,促进剂确保了交联反应的反应动力学的大大改善。
[0089]
在选择的反应参数(根据本发明,特别地,低于发泡层中的聚合物的加工温度的温度)下,在不存在促进剂的情况下,交联反应将不进行,或者将仅以不足的速率进行。例如,用作用于聚丙烯酸酯的交联剂的许多环氧化物固有地相对缓慢地反应,从而在没有促进剂的情况下不产生令人满意的交联结果。
[0090]
质子供体、特别地羧酸和/或羧酸基团和/或其去质子化衍生物在本发明的意义上不计为促进剂。
[0091]
psa层中的促进剂的存在确实也具有缺点。例如,特别地,含氮促进剂例如胺作为氧化过程的结果而随着时间趋向于黄化,使得这种促进剂体系特别不太适合于或不适合于特别地待用于光学目的的透明psa或多层压敏胶带。而且,碱性或酸性促进剂也可随着时间而导致粘合基材的腐蚀。
[0092]
因此,在本发明中,目的是采用反应性足以使其不必掺混任何促进剂的前述共价交联剂和配位交联剂实现psa层的热交联,尤其是与空气接触的psa层的热交联。因此,优选地,本发明的胶带的psa层在不存在促进剂的情况下交联和/或不含促进交联的物质。不存在促进剂在此特别地涉及外部添加的促进剂,换句话说,不共聚和/或引入到聚合物框架中的促进剂。然而,更优选地,psa层既不包含外部添加的促进剂,也不包含共聚的促进剂,且特别地它们根本不含促进剂。
[0093]
如果例如psa被施加到尚未交联的发泡层,或者所述层以特定方法共同成形,则发泡层和一个或两个psa层可同时热交联。
[0094]
然而,各个层也可在单独的过程中进行热交联,如果例如psa被涂覆在已经热交联的发泡层上,然后进行热交联的话,或者如果psa在其它地方(例如,在临时载体材料上)成
形和热交联,然后被层压到已经交联的发泡层上的话。为此,使发泡层和/或psa层经受化学和/或物理预处理可为特别有利的:例如,电晕和/或等离子体处理和/或反应性电晕和/或反应性等离子体处理(使用气体例如氮气、氧气、氟和/或其它)和/或火焰处理。
[0095]
本发明的单面或双面胶带、特别地三层胶带可如ep 1 802 722 a1中对于三层/多层体系所阐述的那样生产。其中描述的生产和涂覆方法也可类似地用于本说明书中描述的胶带;因此,ep 1 802 722 a1的公开内容明确地包括在本公开内容中。这同样适用于ep 1 802 722 a1中的产品构造的描述。
[0096]
用微气球发泡以产生发泡层优选地根据ep 2 414 143 a1和de 10 2009 015 233 a1中描述的方法进行。
[0097]
本发明的胶带的发泡层可被认为是非常高粘度的液体,其在压力载荷下呈现出流动行为(也称为“蠕变”)。粘弹性组合物通常仅通过重力、换言之在由其固有重量产生的载荷下具有或多或少地缓慢流动的能力,特别地流到基材上/润湿基材的能力。然而,至少,这种效果发生在额外的外部压力下。例如,由将胶带压在基材上产生的压力增加可显著促进这种行为。
[0098]
粘弹性组合物所具备的另一能力是,在缓慢暴露于力下放松(释放)作用在它们上的力。因此,它们能够将力消散为振动和/或形变,其也可(至少部分地)为可逆的,且因此能够“缓冲”作用力并且经常能够防止机械破坏,或至少减少这样的破坏或延迟破坏的开始时间。在非常快速的作用力的情况下,粘弹性组合物典型地呈现出弹性行为,换句话说,完全可逆的形变的行为,并且超过组合物的弹性的力可导致破裂。
[0099]
与这些相反的是这样的弹性材料,其即使在缓慢暴露于力下也呈现出所描述的弹性行为。弹性行为根本上对于润湿具有不利结果。尽管有明显的弹性行为,但本发明的psa层在快速暴露于力下也总体上主要呈现出明显的粘弹性行为;特别地,在长的时间尺度中,它们的行为趋向于为粘性的,像流体一样,从而实现了最佳的且特别地迅速的润湿。
[0100]
本发明的胶带、特别地双面带通常地且在上述实施方式中具有一系列特别的优点:
[0101]
作为热交联的结果,胶带在其整个层中没有交联分布。通过光化辐射(紫外线辐射,电子束)交联的粘弹性层或psa层在相应的整个交联的层中呈现出交联分布。热交联的组合物层不呈现出这种行为,因为热量能够均匀地穿透层。
[0102]
凭借根据本发明的共价交联剂和配位交联剂的组合,与使用其它交联剂交联的体系相比,热交联的psa具有更高的粘合力和/或更迅速地达到最终粘合力以及具有更好的热剪切强度。该发现对于本发明的胶带非常重要。在使用发泡的基于聚(甲基)丙烯酸酯的载体并且使其在至少一侧上配备有根据本发明交联的psa的情况下,在该胶带侧上的粘合力以及润湿行为都比在如下体系的情况下更好:
[0103]
‑
所述体系具有在更具弹性的聚合物载体(常规泡沫载体例如基于pe的泡沫载体)上的相应的psa,或
[0104]
‑
所述体系具有相同的泡沫载体,但具有不同的(虽然显著更具粘性)的psa。
[0105]
本发明的胶带的粘合力不仅由psa外层决定,而且同样地由发泡层决定,从而整个体系对于出色的胶粘剂性质是重要的。因此,本发明的胶带所基于的构思包括相对软的泡沫层与以共价和配位两种方式交联的psa层的组合。结果,通过这两个层的相互作用,胶粘
剂性能与在具有不同(以另外的方式)交联的psa层或具有弹性载体的胶带的情况下相比显著更好。
[0106]
除了到目前为止所描述的层之外,本发明的胶带还可包括另外的层,从而形成具有大于3的层序列的多层体系。如果在该情况下,发泡层直接或至少间接地配备有本发明的psa层,则为有利的,因为在该情况下,实现了上述技术胶粘剂优点。更优选地,本发明的胶带由发泡层和根据本发明交联的一个或两个psa层组成。
[0107]
本发明的胶带的特征是,它们可作为非常厚的产品生产,所述非常厚的产品也具有非常高的粘合力。这样的产品见于例如在需要用于补偿不平整性或空腔的胶粘粘合中、在建筑业中或在汽车工业中的应用中。
[0108]
由于发泡层的良好的弛豫行为,本发明的胶带适合于吸收力例如机械应力、冲击等以及消散这样的力的能量。因此,本发明的胶带特别适合于其中需要冲击阻尼和/或振动阻尼效果的应用,如例如在电子产品中的易碎制品的粘合中。当要将具有不同热膨胀系数的材料彼此粘合时采用本发明的胶带是特别的优点,因为本发明的胶带凭借它们的弛豫能力而能够消散由彼此粘合的制品或表面的不同膨胀行为产生的应力。相反,当粘合的制品在其膨胀行为方面大大不同时,常规胶带经常失效,随之而来的是粘合位点的弱化或甚至破裂。
[0109]
本发明的胶带可以几微米至几百微米的惯常胶带厚度生产,但更有利地以超过300μm、例如500μm或更大、1000μm或更大、1500μm或更大、2000μm或更大、或者甚至3000μm或更大的厚度生产。也可实现甚至更厚的产品。
[0110]
在本发明的胶带中,发泡层优选地具有300
‑
2500μm、更优选地400
‑
2400μm的层厚,且所述至少一个psa层优选地具有40
‑
150μm、更优选地50
‑
100μm的层厚。
[0111]
本发明的胶带也特别适合于车辆上的装饰品、徽章和挡泥板的粘合和固定。如果需要,这些表面也可在粘合之前用底漆处理,以进一步提高粘合强度。
[0112]
本发明的胶带出色地适用于其的其它应用领域是,例如,建筑物的建造或扩建、建筑物的装配、以及建筑业(内部和外部二者)、diy行业、建模、家具制造、船舶建造和飞机建造、电子和电气工业(例如,由于良好的热稳定性,也用于消费者电子、白色家电、棕色家电、甚至红色家电)以及用于交通(道路标志等)。
实施例
[0113]
测量方法:
[0114]
固含量(测量方法a1):
[0115]
固含量是聚合物溶液中的非可挥发性成分的分数的量度。其以重量分析的方式测定,其中将溶液称重,然后在120℃下在烘箱中将可挥发组分蒸发蒸发2小时,再次将残余物称重。
[0116]
k值(根据fikentscher)(测量方法a2):
[0117]
k值是高分子化合物中的平均分子尺寸的量度。为了测量,制备1%强度(1g/100ml)的甲苯聚合物溶液,并且使用vogel
‑
ossag粘度计测定其动力学粘度。在按照甲苯的粘度标准化之后,获得相对粘度,并且可用于根据fikentscher(polymer 8/1967,381及以后)计算k值。
[0118]
凝胶渗透色谱法gpc(测量方法a3):
[0119]
本说明书中的重均分子量mw和多分散性pd的数据基于通过凝胶渗透色谱法的测定。对已经历澄清过滤的100μl样品(样品浓度4g/l)进行测定。使用的洗脱液是具有0.1体积%三氟乙酸的四氢呋喃。测量在25℃下进行。使用的预柱是pss
‑
sdv柱,5μ,id 8.0mm
·
50mm。使用如下的柱进行分离:pss
‑
sdv,5μ,以及和各自具有id 8.0mm x 300mm(来自polymer standards service的柱;使用shodex ri71差示折射仪检测)。流速为1.0ml/分钟。对照pmma标准物进行校准(聚甲基丙烯酸甲酯校准)。
[0120]
由涂覆重量和层厚的密度测定(测量方法a4):
[0121]
涂覆的自胶粘剂组合物的每单位体积的重量或密度ρ由单位面积重量对相应的层厚的比率确定:
[0122][0123]
ma=涂覆重量/单位面积重量(没有衬垫重量),以[kg/m2]计
[0124]
d=层厚(没有衬垫厚度),以[m]计
[0125]
该方法产生原始密度。
[0126]
该密度测定特别适合于测定包括多层产品在内的成品的总密度。
[0127]
对钢的90
°
粘合力
‑
开放侧和加衬侧(测量方法m1):
[0128]
对钢的粘合力在23℃ /
‑
1℃温度和50% /
‑
5%相对空气湿度的测试条件下测定。将试样修整到20mm的宽度并粘附到钢板。在测量之前,将钢板清洁和调节。这通过如下完成:首先用丙酮擦拭该板,然后将其留置在空气中5分钟以使溶剂蒸发。
[0129]
然后用50μm铝箔将三层组件的远离测试基材的侧加衬,由此在测量期间防止试样拉伸。随后将测试试样辊压到钢基材上。这通过如下完成:用2kg辊以10m/分钟的辊压速度在该带上来回辊压5次。在辊压施用之后,立即将钢板插入特定支架中,该特定支架允许使试样以90
°
的角度被向上拉出。使用zwick拉伸测试机测量粘合力。当将加衬侧施加到钢板时,首先将三层组件的开放侧层压到50μm铝箔,除去剥离材料,并且将试样粘附到钢板,类似地辊压,并进行测量。
[0130]
两侧(开放侧和加衬侧)的测量结果以n/cm作为来自三次测量的平均值报告。
[0131]
保持力
‑
开放侧和加衬侧(测量方法m2):
[0132]
在23℃ /
‑
1℃温度和50% /
‑
5%相对空气湿度的测试条件下制备试样。将测试试样修整到13mm并粘附到钢板。粘合面积为20mm
·
13mm(长度
·
宽度)。在测量之前,将钢板清洁和调节。这通过如下完成:首先用丙酮擦拭该板,然后将其留置在空气中5分钟以使溶剂蒸发。在粘合之后,将开放侧用50μm铝箔增强,并且使用2kg辊来回辊压2次。然后将带环安装在三层组件的突出端上。随后将该结构体悬挂在合适的装置处并且用规定的重物加载;在各实施例中指定重物。悬挂装置使得重物以179
°
/
‑1°
的角度加载样品。这确保了三层组件不能从板的下边缘剥离。以分钟报告测量的保持力(其为在试样的悬挂和其掉落之间的时间)并且其对应于来自三次测量的平均值。为了测量加衬侧,首先将开放侧用50μm铝箔增强,除去剥离材料,并且与所描述的方式类似地进行对测试板的粘附。测量在标准条件(23℃,55%湿度)下进行。
[0133]
标示牌测试(npt)(测量方法m3):
[0134]
将2cm宽、15cm长和0.5mm厚的铝测试条用丙酮洗涤,并且在23℃ /
‑
1℃温度和50% /
‑
5%相对空气湿度的条件下留置5分钟。然后将铝条纵向施加到胶带试样。然后将突出的胶带切断,使得该带的端部与铝板齐平。将20cm长、2.5cm宽、和3mm厚的聚碳酸酯板(pc板)用乙醇洗涤,并且在23℃ /
‑
1℃温度和50% /
‑
5%相对空气湿度的条件下留置120分钟。由铝板和胶带形成的组件居中地粘贴在pc板上并由此制造测试试样。通过如下确保规定的粘合:使用4kg辊在组件上来回辊压5次,然后将其留置72小时。
[0135]
将具有粘贴在其上的由胶带试样和铝条组成的组件的pc板夹在33
°
npt框架中,该框架的构造示于图1和2中。图1显示框架的横截面,其具有弯曲的金属板40。用a、b和c标示的尺寸的定义如下:
[0136]
a=211mm;
[0137]
b=28mm;
[0138]
c=6mm。
[0139]
角度α的大小为33
°
。
[0140]
如图2另外示出的,除了拱形金属板40之外,该框架主要包括安装在侧面上的固定轨道50和调节螺钉60。
[0141]
然后,将pc板在横向方向上以这样的方式夹在该框架中,使得其端部通过固定轨道与该框架齐平地被固定,其中粘贴的铝板明显地朝上而不固定。将该框架在50℃下的烘箱中储存。在1、24和48小时之后,分别以相对于pc板90
°
的角度测量铝测试条的两端之间的距离。测量结果是两个测量距离的总和并以mm报告。进行重复的测定,并计算平均值。
[0142]
测量的结果解释如下:
[0143]
≤5mm:有利的
[0144]
<10mm:令人满意的
[0145]
10
–
16mm:仍然不足的
[0146]
≥17mm:不足的。
[0147]
所使用的可商购获得的材料
050 000g/mol。k值:50.5。
[0153]
聚丙烯酸酯psa 2(pa2):
[0154]
将100l常规用于自由基聚合的玻璃反应器装填4.0kg丙烯酸、12.0kg ba、24.0kg丙烯酸2
‑
丙基庚酯pha以及26.7kg丙酮/汽油60/95(1:1)。在搅拌下使氮气通过反应器45分钟之后,将反应器加热至58℃并且添加30gaibn。然后将外部加热浴加热至75℃,并且恒定地在该外部温度下进行反应。在1小时的反应时间后,添加另外的30g aibn。在4小时和8小时后,分别用10kg丙酮/汽油60/95(1:1)混合物稀释该批料。为了减少残留的引发剂,在8小时后和在10小时后分别添加90g的16。在24小时的时间后停止反应,并且将批料冷却至室温。然后将聚丙烯酸酯与交联剂共混,用丙酮稀释至30%的固含量,然后以与对于pa1相同的方式进行涂覆和干燥。涂覆重量为50g/m2。通过gpc的摩尔质量(测量方法a3):mn=25 700g/mol;mw=891 000g/mol。k值:49.8。
[0155]
聚丙烯酸酯psa 3(pa3):
[0156]
将100l常规用于自由基聚合的玻璃反应器装填3.2kg丙烯酸、4.8kg丙烯酸异冰片酯(iboa)、17.0kg eha、15.0kg ba以及26.7kg丙酮/汽油60/95(1:1)。在搅拌下使氮气通过反应器45分钟之后,将反应器加热至58℃并且添加30g aibn。然后将外部加热浴加热至75℃,并且恒定地在该外部温度下进行反应。在1小时的反应时间后,添加另外的30g aibn。在4小时和8小时后,分别用10kg丙酮/汽油60/95(1:1)混合物稀释该批料。为了减少残留的引发剂,在8小时后和在10小时后分别添加90g的双(4
‑
叔丁基环己基)过氧化二碳酸酯。在24小时的时间后停止反应,并且将批料冷却至室温。然后将聚丙烯酸酯与交联剂共混,用丙酮稀释至30%的固含量,然后以与对于pa1相同的方式进行涂覆和干燥。涂覆重量为50g/m2。通过gpc的摩尔质量(测量方法a3):mn=25 100g/mol;mw=1 080 000g/mol。k值:51.1。
[0157]
聚丙烯酸酯psa 4(pa4):
[0158]
将100l常规用于自由基聚合的玻璃反应器装填4.0kg丙烯酸、18.0kg eha、18.0kg ba以及26.7kg丙酮/汽油60/95(1:1)。在搅拌下使氮气通过反应器45分钟之后,将反应器加热至58℃并且添加30g aibn。然后将外部加热浴加热至75℃,并且恒定地在该外部温度下进行反应。在1小时的反应时间后,添加另外的30g aibn。在4小时和8小时后,分别用10kg丙酮/汽油60/95(1:1)混合物稀释该批料。为了减少残留的引发剂,在8小时后和在10小时后分别添加90g的双(4
‑
叔丁基环己基)过氧化二碳酸酯。在24小时的时间后停止反应,并且将批料冷却至室温。然后将聚丙烯酸酯与交联剂共混,用丙酮稀释至30%的固含量,然后以与对于pa1相同的方式进行涂覆和干燥。涂覆重量为50g/m2。通过gpc的摩尔质量(测量方法a3):mn=21 000g/mol;mw=1 05 000g/mol。k值:50.0。
[0159]
表1列出了本发明实施例b1
‑
b6和对比例vb7
‑
vb11的组成。
[0160]
表1:实施例b1
‑
b6、b11、b12以及对比例vb7
‑
vb10
[0161][0162][0163]
1)
对于每g的聚合物,共价交联剂的官能团或配位交联剂的结合位点的mmol
[0164]
2)
al螯合物=乙酰丙酮铝
[0165]
ii.用于聚丙烯酸酯泡沫体vt1和vt2以及压敏胶带实施例mt1至mt14的起始聚合物的制备
[0166]
下面描述的是起始聚合物的制备,其常规地通过自由基聚合在溶液中制备。
[0167]
基础聚合物p1
[0168]
将常规用于自由基聚合的反应器装填3.0kg丙烯酸、30.0kg eha、67.0kg ba和66kg丙酮/异丙醇(96:4)。在搅拌下使氮气通过反应器45分钟之后,将反应器加热至58℃并且添加50g67。然后将外部加热浴加热至75℃,并且恒定地在该外部温度下进行反应。在1小时后,添加另外的50g67,并且在4小时后,用20kg丙酮/异丙醇混合物(96:4)稀释该批料。在5小时后和在7小时后,分别用150g16进行再引发,并且用23kg丙酮/异丙醇混合物(96:4)进行稀释。在22小时的反应时间之后,停止聚合且将批料冷却至室温。聚丙烯酸酯具有75.1的k值、50.2%的固含量以及mn=91 900g/mol和mw=1 480 000g/mol的平均分子量。
[0169]
基础聚合物p2
[0170]
将常规用于自由基聚合的反应器装填10.0kg丙烯酸、45.0kg eha、45.0kg ba和66kg丙酮/异丙醇(94:6)。在搅拌下使氮气通过反应器45分钟之后,将反应器加热至58℃并
且添加50g67。然后将外部加热浴加热至75℃,并且恒定地在该外部温度下进行反应。在1小时后,添加另外的50g67,并且在4小时后,用25kg丙酮/异丙醇混合物(94:6)稀释该批料。在5小时后和在7小时后,分别用150g16进行再引发,并且用23kg丙酮/异丙醇混合物(94:6)进行稀释。在22小时的反应时间之后,停止聚合且将批料冷却至室温。聚丙烯酸酯具有49.1的k值、49.5%的固含量以及mn=21 900g/mol和mw=890 000g/mol的平均分子量。
[0171]
过程1:热熔性psa的浓缩/制备:
[0172]
借助于单螺杆挤出机(浓缩挤出机,berstorff gmbh,germany)使基础聚合物p基本上不含溶剂(残留溶剂含量≤0.3重量%)。浓缩基础聚合物的参数如下:螺杆转速为150rpm,电动机电流为15a,并且实现58.0kg/小时液体的通量。对于浓缩,在三个不同的圆顶处施加真空。降低的压力分别在20毫巴和300毫巴之间。浓缩的热熔体p的出口温度为约115℃。在该浓缩步骤后的固含量为99.8%。
[0173]
过程2:本发明的胶带的生产,与用于热交联的交联剂
‑
促进剂体系的共混,和涂覆
[0174]
在对应于图3中的图示的实验单元中进行发泡。
[0175]
将浓缩的基础聚合物p通过过程1在进料器挤出机1中熔融并且将其通过该挤出机以聚合物熔体的形式经由可加热软管11输送到来自entex(bochum)的行星辊挤出机2(pwe)中(使用的pwe特别地具有可彼此独立地加热的四个模块t1、t2、t3和t4)。经由计量开口22,可供应另外的添加剂或填料例如颜色糊料。在点23处,添加交联剂。将所有组分混合以形成均匀的聚合物熔体。
[0176]
借助于熔融泵24a,将聚合物熔体转移到双螺杆挤出机3(来自berstorff)(进料位置33)。在位置34处,添加促进剂组分。随后在175毫巴的压力下在真空圆顶v中使混合物整体上不含所有气体夹杂物(参见以上对于无气状态的标准)。在真空区的下游,在螺杆上,存在泡罩b,其允许随后的区段s中的压力形成。通过适当地控制挤出机转速和熔体泵37a,在泡罩b和熔融泵37a之间在区段s中形成大于8巴的压力;在计量位点35处,添加微气球混合物(根据表2中的细节将微气球嵌入分散助剂中),并且借助于混合元件将其均匀地引入预混物中。将得到的熔融混合物转移到模头5中。
[0177]
在从模头5离开后,换句话说在压力下降后,引入的微气球经历膨胀,其中压力下降导致聚合物组合物的低剪切、更特别地无剪切冷却。这产生发泡的psa,其随后被涂覆在两个剥离材料之间,所述剥离材料可在除去之后再次使用(工艺衬垫),并且借助于辊压压延机4将psa成形为幅材。
[0178]
为了改善来自本发明的实施例和对比例的psa对成形的聚丙烯酸酯泡沫体的锚定,psa以及泡沫体均被电晕预处理(来自vitaphone,denmark的电晕单元,70w
·
分钟/m2)。在生产三层组件之后,该处理导致改善的对聚丙烯酸酯泡沫载体层的化学连接。
[0179]
在通过涂覆单元期间的幅材速度为30m/分钟。
[0180]
在离开辊隙后,除去任何抗胶粘性载体,并且将完成的三层产品与剩余的第二抗胶粘性载体一起卷绕。
[0181]
表2:聚丙烯酸酯泡沫体vt1和vt2
[0182][0183]
密度:测量方法a4
[0184]
以下呈现的是生产本发明和对比例的胶带的具体实施例,本发明的胶带包括聚丙烯酸酯泡沫载体vt1和vt2与本发明的psa实施例b1
–
b6和b11
–
b12,具有50g/m2的双面涂覆重量,并且对比例的胶带包括聚丙烯酸酯泡沫载体vt1和vt2与非本发明的psa实施例vb7
–
vb10,同样具有50g/m2的双面涂覆重量。
[0185]
表3:具有1000μm的总厚度的包括聚丙烯酸酯泡沫载体vt1或vt2的三层压敏胶带mt1
‑
mt14的对钢和abs的粘合力以及剥离行为
[0186]
[0187]
comp.=对比例,psa=压敏胶粘剂,vt=泡沫载体,f.s.=泡沫体分裂
[0188]
由表3中的粘合力测量明晰,本发明的psa带非常快速地粘附到钢并达到其最大粘合力,或导致聚丙烯酸酯泡沫载体的分裂。此外,所有实施例同样对abs呈现出良好的剥离粘附值。
[0189]
相反,如果仅使用共价交联剂或配位交联剂,则达到平衡状态下的剥离粘附值常常需要更长时间,并且对abs的粘合力略微较低。在实施例mt8中,共价交联剂的量以及聚合物与对于实施例mt1的相同;发现,省略可逆的配位交联剂损害剥离行为。类似地,在实施例mt10中,配位交联剂中的结合位点的总数与实施例mt1相当;明晰的是,粘合力降低。在实施例mt12
‑
14中,共价基团对配位交联剂的结合位点的比率低于或高于优选比率。这对于剥离行为或对于粘合力水平没有不利后果。
[0190]
表4:具有1000μm的总厚度的三层psa带mt1
–
mt14的保持力时间和标示牌测试结果
[0191][0192]
ssz=保持力时间23
°
和70℃=测量方法m2,a:胶粘失效,co:内聚失效;npt=标示牌测试=测量方法m3
[0193]
压敏胶带之间的差异最佳地可见于标示牌测试中(表4)。在此明晰的是,单独使用共价交联剂特别地可导致在标示牌测试中的明显的边缘提升(翘起)(mt8,mt9)。当以不那么优选的官能团对结合位点的比率使用共价交联剂和配位交联剂的组合(mt12
–
mt14)时,可在较小程度上看到类似的行为,其中在此另外地可明晰泡沫载体的影响。与略微较软的载体vt1(mt12)相比,由于较高的丙烯酸浓度而较硬的载体vt2(mt13)导致在1小时后的相对差的初始值。另一方面,如果仅使用配位交联剂(mt10,mt11),则所得的标示牌测试结果与本发明的交联剂体系的那些大致相当;然而,热剪切强度显著较差。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。