1.本技术涉及荧光标记领域,更具体地说,它涉及一种新型噻唑偶氮染料及其制备方法及其应用。
背景技术:
2.荧光分析法因其具有灵敏度高、选择性好、信息量丰富、检测限低、试样量少、操作简单等显著优点,故荧光分析法在环境检测、食品分析及医药分析等领域得到广泛应用,但是自身具有内源荧光的药物相对较少,导致大多数的药物无法用直接荧光法进行定量分析,而是要采用荧光猝灭法进行检测。
3.荧光猝灭法是利用某种无荧光特性或荧光特性微弱的待测物质对某一荧光物质的猝灭作用,从而依据荧光降低程度与待测物浓度之间的关系,来进一步研究待测物浓度或者含量的荧光分析法。这类分析方法操作相对容易,不需要复杂的仪器,且与直接荧光法相比,其灵敏度更高,选择性更好,在生物分析和药物分析中日益引起重视。
4.荧光猝灭是指由于荧光物质分子与溶剂或溶质分子之间所发生的导致荧光强度下降的物理或化学作用的过程。相互作用引起的荧光降低的现象,这些会引起荧光的猝灭的物质称为荧光猝灭剂。
5.针对上述相关技术,发明人认为现有的荧光猝灭剂合成工艺复杂且收率、纯度不高,例如,猝灭剂pfo/ppv的制备:在反应瓶中加入1,4
‑
二乙烯基苯,9,9
‑
二辛烷基
‑
2,7
‑
二溴芴,三(4
‑
溴苯基)胺,pd(oac)2,pph3,n,n
′‑
二甲基甲酰胺,抽排3次除去空气后,用针筒滴加除水除氧的三乙胺,100℃搅拌反应48h,反应完毕后冷却到室温倒入甲醇和hcl混合溶液中沉淀,过滤得到固体,用氯仿溶解,过滤掉不溶物,滤液浓缩后再用甲醇进行沉淀.反复沉淀3次,真空干燥,得黄色固体,且产率仅有66%。合成步骤复杂且产率低使得荧光猝灭剂的选择存在限制,因此,还有改善的空间。
技术实现要素:
6.为了使得荧光猝灭剂的合成工艺简单,收率、纯度高,本技术提供一种新型噻唑偶氮染料及其制备方法和应用。
7.第一方面,本技术提供一种新型噻唑偶氮染料的制备方法,采用如下的技术方案:
8.一种新型噻唑偶氮染料的制备方法,包括以下步骤:
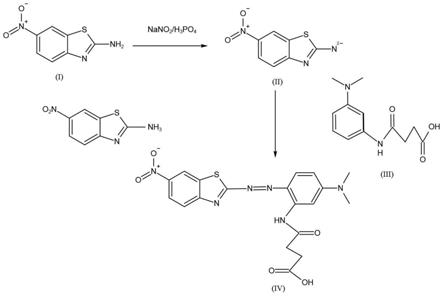
9.步骤1),取化合物i分散在磷酸溶液中,待固体完全溶解后冷却至0℃以下,加入含亚硝酸根的盐溶液,在0℃以下反应1.5~3h,获得化合物ii;
10.步骤2),将化合物iii溶解于醇类溶剂中,冷却至0℃以下,获得溶解液备用;
11.步骤3),将化合物ii倾倒入溶解液中,在0℃以下反应1.5~3h,然后过滤、干燥,获得新型噻唑偶氮染料;
12.所述化合物i与化合物iii的摩尔比为1:0.8~1.2;
13.所述化合物i的结构式为式i:
14.所述化合物ii的结构式为式ii:
15.所述化合物iii的结构式为式iii:
16.由化合物i和化合物iii经过上述制备方法,通过两步反应即可制得新型噻唑偶氮类染料,并且产率达到74.3%以上,纯度达到97.5%以上,得到的目标产物能在荧光共振能量转移(fret)中用作猝灭剂,为荧光猝灭领域提供了一个新的猝灭剂,从而拓宽了荧光猝灭技术在生物分析和药物分析领域的应用。
17.优选的,r1、r2、r3为氢、烷基或卤素,r4为烷基。
18.术语“烷基”在本技术中按本领域普通技术人员已知的含义使用,指仅由碳和氢原子组成的一价残基,烷基形成具有通式c
n
h
2n 1
的同类系列。烷基可为直链或支链烷基,例如烷基可为仲烷基或叔烷基,仲烷基为支链,具有与两个碳残基连接的中心碳原子;叔烷基为支链,具有与3个碳残基连接的中心碳原子。r1、r2、r3、r4中的烷基可为例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、2
‑
戊基、3
‑
戊基、2
‑
甲基丁基、3
‑
甲基丁基、3
‑
甲基丁
‑2‑
基、2
‑
甲基丁
‑2‑
基、2,2
‑
二甲基
‑
丙基、正己基、2
‑
甲基
‑
戊基、3
‑
甲基
‑
戊基、2
‑
二甲基
‑
丁基、3
‑
二甲基
‑
丁基、4
‑
二甲基
‑
丁基、2,3
‑
二甲基丁基、2,4
‑
二甲基丁基或3,4
‑
二甲基丁基、2
‑
乙基丁基、3
‑
乙基丁基、2
‑
甲基
‑
戊基或3
‑
甲基
‑
戊基。
19.更优选的,所述r1、r2、r3、r4为甲基、乙基、正丙基、异丙基或叔丁基。
20.最优选的,所述r1为氢,r2、r3为甲基,r4为乙基。
21.优选的,所述步骤1)中化合物i溶解在磷酸溶液中时边加热边溶解。
22.化合物i在加热状态下加速了分子之间碰撞的概率,使得化合物i更易与溶解于磷酸溶液中,减少了制备噻唑偶氮染料的制备时间,减少了因制备时间长而产生的一些副反应,从而使得制备噻唑偶氮染料的产率、纯度得以进一步提高。
23.优选的,步骤1)中分2~5次以10~30g/min加入含亚硝酸根的盐溶液。
24.为了使化合物i与亚硝酸根离子充分反应,通过缓慢加入含有亚硝酸根离子的盐溶液,使得亚硝酸离子在反应溶液中有足够的结合位点能够充分地反应,减少因一次性加入过多的化合物使反应不充分而产生副反应,从而使得制备噻唑偶氮染料的产率、纯度得以进一步提高。
25.优选的,所述步骤3)中将化合物ii以8~15g/min的速度缓慢倒入溶解液中。
26.以8~15g/min的速度将化合物iii倒入溶解液中,使得化合物iii与溶解液中的化合物iii能够成分反应而不易产生副产物,从而进一步提高了制备噻唑偶氮染料的产率、纯度。
27.第二方面,本技术提供一种新型噻唑偶氮染料,采用如下的技术方案:
28.一种新型噻唑偶氮染料,结构式如下:
[0029][0030]
其中r1、r2、r3为氢、烷基或卤素,r4为烷基。
[0031]
本技术制备的新型噻唑偶氮染料同时携带有吸电子的硝基和羧基,在作为荧光猝灭的受体时,能级远低于供体的能级,使能量发生有效的转移,通过羧基的修饰升高了噻唑偶氮染料的还原电位,使得新型偶氮染料具有激发态分子的特征,因为激发态分子壁基态分子更强的电子给体,因此更容易与其他物质的分子发生电荷转移作用,起到荧光猝灭的效果,从而可以用来标记蛋白质。
[0032]
第三方面,本技术提供一种新型噻唑偶氮染料在荧光共振能量转移(fret)中用作猝灭剂。
[0033]
综上所述,本技术具有以下有益效果:
[0034]
1、由于本技术采用由化合物i和化合物iii经过两步反应即可制得新型噻唑偶氮类染料,并且产率达到74.3%以上,纯度达到97.5%以上,拓宽了荧光猝灭技术在生物分析和药物分析领域的应用。
[0035]
2、本技术中优选采用以8~15g/min的速度将化合物ii倒入溶解液中,使得化合物ii与溶解液中的化合物iii能够成分反应而不易产生副产物,从而进一步提高了制备噻唑偶氮染料的产率、纯度。
[0036]
3、本技术的新型噻唑偶氮染料应用于荧光猝灭领域具有高选择性、高准确定的优点。
附图说明
[0037]
图1本技术实施例1中的制备新型噻唑偶氮染料的合成路线图。
[0038]
图2是实施例1中制备的新型噻唑偶氮染料的质谱图:lrms(esi
‑
):m/zcalcdforc
19
h
18
n6o5s[m
‑
h]
‑
442.1;und442.2。
[0039]
图3实施例1制备的新型噻唑偶氮染料的hplc图:1hnmr(400mhz,cdcl3)δ11.90(s,1h),8.62(s,1h),8.29(d,1h),8.05(d,1h),7.16(m,1h),6.65
‑
6.75(m,2h),2.96(s,6h),
2.68(t,2h),2.47(t,2h)ppm。
具体实施方式
[0040]
以下结合附图和实施例对本技术作进一步详细说明。
[0041]
实施例
[0042]
实施例中的化合物i的结构式为:
[0043]
化合物ii的结构式为:
[0044]
化合物iii的结构式为:
[0045]
实施例1
[0046]
本实施例公开一种新型噻唑偶氮染料的制备方法,包括以下步骤:
[0047]
步骤1),取化合物i11.56g分散在装有350g质量浓度为85%磷酸溶液的烧瓶中,待固体完全溶解后进行冰浴,冷却至0℃后加入4.9g亚硝酸钠溶液,在0℃反应3h,获得化合物ii;
[0048]
步骤2),将16.8g化合物iii溶解于装有甲醇的烧瓶中,冰浴冷却至0℃,获得溶解液备用;
[0049]
步骤3),将化合物ii倾倒入装有溶解液的烧瓶中,在0℃的条件下反应3h,然后过滤,过滤后的滤饼用水洗涤,在烘箱干燥,获得8.59g黑红色晶体状的新型噻唑偶氮染料。
[0050]
实施例2
[0051]
本实施例公开一种新型噻唑偶氮染料的制备方法,包括以下步骤:
[0052]
步骤1),取化合物i11.56g分散在装有320g质量浓度为85%磷酸溶液烧瓶中,待固体完全溶解后进行冰浴,冷却至0℃后加入4.1g亚硝酸钠溶液,在0℃反应2h,获得化合物ii;
[0053]
步骤2),将14g化合物iii溶解于装有甲醇的烧瓶中,冰浴冷却至0℃,获得溶解液备用;
[0054]
步骤3),将化合物ii倾倒入装有溶解液的烧瓶中,在0℃的条件下反应2h,然后过
滤,过滤后的滤饼用水洗涤,在烘箱干燥,获得9.09g黑红色晶体状的新型噻唑偶氮染料。
[0055]
实施例3
[0056]
本实施例公开一种新型噻唑偶氮染料的制备方法,包括以下步骤:
[0057]
步骤1),取化合物i11.56g分散在装有300g质量浓度为85%磷酸溶液的烧瓶中,待固体完全溶解后进行冰浴,冷却至0℃后加入3.3g亚硝酸钠溶液,在0℃反应1.5h,获得化合物ii;
[0058]
步骤2),将11.2g化合物iii溶解于装有甲醇的烧瓶中,冰浴冷却至0℃,获得溶解液备用;
[0059]
步骤3),将化合物ii倾倒入装有溶解液的烧瓶中,在0℃的条件下反应1.5h,然后过滤,过滤后的滤饼用水洗涤,在烘箱干燥,获得9.00g黑红色晶体状的新型噻唑偶氮染料。
[0060]
实施例4
[0061]
与实施例2的区别在于:步骤1)至步骤3)中的冷却温度均为
‑
5℃,获得9.10g黑红色晶体状的新型噻唑偶氮染料。。
[0062]
实施例5
[0063]
与实施例4的区别在于:步骤1)中反应物i溶解在磷酸溶液中时,用电加热套边加热边溶解,获得9.14g黑红色晶体状的新型噻唑偶氮染料。
[0064]
实施例6
[0065]
与实施例2的区别在于:步骤1)中分2次以30g/min加入含亚硝酸根的盐溶液,获得9.17g黑红色晶体状的新型噻唑偶氮染料。
[0066]
实施例7
[0067]
与实施例2的区别在于:步骤3)中将化合物iii以8g/min的速度缓慢倒入装有溶解液的烧瓶中,获得9.12g黑红色晶体状的新型噻唑偶氮染料。
[0068]
实施例8
[0069]
与实施例5的区别在于:
[0070]
步骤1)中分5次以10g/min加入含亚硝酸根的盐溶液。
[0071]
步骤3)中将化合物iii以15g/min的速度缓慢倒入装有溶解液的烧瓶中,获得9.30g黑红色晶体状的新型噻唑偶氮染料。
[0072]
检测试验
[0073]
1、产率=新型噻唑偶氮染料的质量/(化合物i 化合物iii的质量)
·
%。
[0074]
2、利用高效液相色谱法测定新型噻唑偶氮染料的纯度。
[0075]
质谱数据:lrms(esi
‑
):m/zcalcdforc
19
h
18
n6o5s[m
‑
h]
‑
442.1;und442.2.详见说明书附图2。
[0076]
核磁共振氢谱数据:1hnmr(400mhz,cdcl3)δ11.90(s,1h),8.62(s,1h),8.29(d,1h),8.05(d,1h),7.16(m,1h),6.65
‑
6.75(m,2h),2.96(s,6h),2.68(t,2h),2.47(t,2h)ppm,详见说明书附图3。
[0077]
表1
[0078]
项目产率/%hplc纯度/%实施例174.397.3实施例278.697.7
实施例377.998.1实施例478.797.8实施例579.198.0实施例679.398.2实施例778.997.9实施例880.598.6
[0079]
根据表1中的数据可得,化合物i和化合物iii通过两步反应即可制得灵敏度更高、选择性更好的新型噻唑偶氮类染料,并且产率高达80.5%,纯度高达98.6%,新型噻唑偶氮类染料能在荧光共振能量转移(fret)中作为猝灭剂,为荧光猝灭领域提供新的猝灭剂,拓宽了荧光猝灭技术在生物分析和药物分析领域的应用,推动了药物质量标准的检测。
[0080]
本具体实施例仅仅是对本技术的解释,其并不是对本技术的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本技术的权利要求范围内都受到专利法的保护。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。