1.本发明属于分子生物学及基因工程相关技术领域,涉及一种天麻超氧化物歧化酶(superoxide dismutase,sod)基因以及在提高天麻抗寒性或促进拟南芥生长中的应用。
背景技术:
2.天麻(gastrodia elata)是与真菌共生的异养多年生草本植物,也是一种名贵传统中药材。天麻素和4
‑
羟基苯甲醇是天麻的主要药效成分,具有防治老年痴呆、阿尔兹海默症、抑郁症、中风以及改善记忆等功效。由于天麻营养丰富,是目前备受青睐的保健食品。近年来,随着生态环境不断破坏和人工采挖,野生天麻资源减少或濒临灭绝。目前天麻供不应求,人工栽培天麻将成为满足市场需求的唯一有效途径。
3.气候条件影响天麻生长发育,影响天麻生长发育的因素有很多,如环境、温度、土壤、采收等,其中温度是影响天麻生长的关键因子;天麻对温度很敏感,当温度过高或过低,都会抑制天麻的生长;天麻生长最适宜温度为 20~25℃;在
‑
4℃以下时天麻易受到冻害死亡而丧失繁殖能力,温度在
‑
4~10℃时则天麻停止生长进入低温休眠期;当土温上升至12~14℃时,天麻的地下块茎开始萌动生长;当温度上升至20℃时,天麻进入迅速生长期,但温度超过30℃时天麻的生长会受到抑制,从而影响产量。
4.蜜环菌是一种以腐生为主的兼寄生真菌,主要寄生于阔叶树的根、茎和叶,分解有机物质如纤维素、半纤维素、木质素的能力强。蜜环菌是促进天麻生长发育的重要菌根真菌,蜜环菌对温度也很敏感;蜜环菌在 6~8℃时开始生长,生长最适温度为 24℃,超过 30℃就停止生长。
5.目前还未见关于天麻超氧化物歧化酶(superoxide dismutase,sod)基因在提高天麻抗寒性或促进拟南芥生长中的报道。
技术实现要素:
6.针对现有技术存在的问题,本发明以不同温度下生长的昭通乌天麻(gastrodia elata)为研究对象,利用转录组数据组装后得到163261356个 clean reads 片段,共有33702个 unigene 被注释到kegg 信号通路数据库,分析发现在所有途径中,富集基因最多的是碳代谢途径,其次是翻译、脂质代谢和植物激素信号传导途径;说明代谢途径、次生代谢产物合成、rna 转运、脂质代谢等的差异基因表达与不同温度下天麻的生长发育的作用机制有关。从转录组数据里筛选出与代谢途径相关的关键基因
‑‑‑
超氧化物歧化酶基因。对差异表达基因超氧化物歧化酶基因进行克隆与功能鉴定;获得一种天麻超氧化物歧化酶(superoxide dismutase,sod)基因,其核苷酸序列如seq id no:1所示,编码如seq id no:2所示的氨基酸序列,本发明天麻超氧化物歧化酶(sod)基因全长720bp,编码 239 个氨基酸残基。
7.本发明另一目的是将上述天麻超氧化物歧化酶基因应用在提高天麻共生菌蜜环菌抗寒性中。
8.为了实现本发明的上述目的,本发明的技术方案如下:1、蜜环菌am02活化(实验室自己分离,保存于实验室冷库中),具体步骤为
①
将蜜环菌菌索取出置于pda平板(马铃薯200g,葡萄糖20g,琼脂18g,磷酸二氢钾3g,硫酸镁1.5g,ddh2o 1l,ph自然,115℃灭菌20min)上,25℃培养6天;
②
待pda培养基上长出白色幼嫩菌索后,将幼嫩菌索边缘的白色菌丝用针头挑于新pda培养基(马铃薯200g,葡萄糖20g,琼脂20g,ddh2o 1l,ph5.5,115℃灭菌20min)培养,此步可重复;
③
至从中央向外缘长出白色菌丝后,挑取边缘的菌丝于液体完全培养基(葡萄糖46g,酵母膏5g,蛋白胨13g,硫酸镁2g,磷酸二氢钾1g,ddh2o 1l,ph 6.5,115℃灭菌20min)中 25℃、150 rpm下避光培养 10d,长出菌球备用;蜜环菌菌材的制备,步骤具体如下:
①
选质地坚硬、耐腐蚀的阔叶树进行蜜环菌菌材的培养;
②
将选用的苹果树锯成小段,放清水中浸泡一夜,清洗干净后置于培养瓶中,121℃下灭菌20min ,灭菌3次以上,以防止木材中的杂菌污染蜜环菌;
③
将蜜环菌用frh
‑
2a 破碎机(金坛市白塔新宝仪器厂产)破碎后倒入灭好菌的木材中,待蜜环菌缠于木材上长出菌索后,说明菌材制备成功备用;2、本发明从云南昭通采集新鲜白头麻(连土一起采回,短暂存于4℃冷库),将从昭通采集回来的置于4℃冷库的第二年生的天麻(白头麻)从土壤里挖出来,用流水清洗天麻块茎表面的泥土残渍,将块茎放75 %乙醇中处理3min,无菌水冲去残留的乙醇,再置于2.5% naclo溶液中浸泡10min,无菌水冲去残留的naclo溶液,用超纯水洗干净并置于灭菌水中洗2
‑
3次,将洗净的白头麻表面水分吸干后,接种于蜜环菌菌材上;分别在4℃冷库、13℃恒温箱(箭麻生长的临界温度)和 23℃培养室(正常生长温度)黑暗培养条件下培养,然后蜜环菌菌索开始侵染昭通乌天麻,给白头麻(母麻)提供营养,使其发育形成箭麻(商品麻),第6d 观察得到23℃下箭麻开始生长,第40d 观察到13℃下箭麻开始生长, 第50d观察4℃下天麻不发芽;天麻培养至50d 时采集不同温度下生长的天麻样品;将13℃母麻、23℃商品麻和 23℃母麻送公司进行转录组测序,从转录组中筛选log2(变化倍数)>2的差异表达基因sod基因;因此取13℃下培养的母麻,采用trizol reagent(invitrogen)法提取天麻的总rna,rna反转录获得cdna,以cdna为模板,采用巢式pcr引物,进行两次巢式pcr,获得250bp以内的未知片段,胶回收后连接到pmd
‑
18t 载体上,转入大肠杆菌dh5α中,测序;将测序结果与靠近已知序列的3’端的引物比对上,再看引物之后的几个碱基是否成功比对,成功比对就说明未知片段比对成功,比对成功后,已知的最后一个碱基开始拼接得一条以atg开始的片段,用dnaman输入拼接得到的序列,查找终止密码子,获得以atg开始,以tag终止的一段cdna全长为720bp;2、构建sod基因原核表达载体将pmd
‑
18t
‑
sod和pet
‑
32a质粒进行酶切,跑胶检测,将pmd
‑
18t
‑
sod质粒酶切的目的条带胶回收,将pet
‑
32a质粒酶切的载体胶回收;原核表达载体连接,取2μlpet
‑
32a
‑
sod质粒用热击法转化感受态细胞bl21(de3),挑单菌落于100μg/ml amp抗性50ml lb液体培养基中,37℃、200 r/min摇床过夜培养;取2μl菌液做pcr检测扩增目的条带是否正确,扩增得到目的sod条带说明,转化bl21大肠杆菌成功;将测序成功的pmd
‑
18t
‑
sod质粒和pet
‑
32a空载质粒进行双酶切,pmd
‑
18t
‑
sod质粒切出两条序列,其中一条序列为720bp(图1a),与sod扩增片段大小吻合;pet
‑
32a空载质粒切出一条序列(图1b);将sod片段和pet
‑
32a空目的载
体胶回收后,连接转化dh5α,提质粒酶切检测(图1c),pet
‑
32a
‑
sod质粒切出两条序列,其中一条序列为720bp,质粒测序结果比对与sod全长序列相似率为100%,说明pet
‑
32a
‑
sod载体构建成功。取2
µ
l pet
‑
32a
‑
sod质粒转入bl21后菌液pcr得到一条720bp大小的条带(图1d ),表明pet
‑
32a
‑
sod原核表达载体构建成功;将转化bl21成功的菌液用iptg诱导蛋白表达,加入1mmol/l iptg分别置于28℃、37℃摇床进行诱导表达;分别取不加iptg(对照)和加iptg诱导表达至0h、2h、4h、6h、8h菌液,进行sds
‑
page(12%分离胶和4%浓缩胶)分析,跑蛋白胶后,用考马斯亮蓝g
‑
250染色液染色,用脱色液脱色1h,脱色后观察重组蛋白sod表达水平,以确定其最佳诱导表达温度及时间;可溶性蛋白检测:将上一步验证得到的诱导最适时间对应的菌液,也就是加入1mmol/l 的iptg诱导剂 28℃和 37℃诱导6h的菌液,离心弃上清,然后向沉淀中加入沉淀等体积的冰上预冷的1
×
pbs 缓冲液,重悬菌体;再用超声破碎机破碎 bl21 细胞(冰上操作)10min,4℃、12000rpm离心2min,将上清移至新离心管中,沉淀用8mol/l 尿素溶解;最后在不加 iptg的上清、沉淀,加iptg 28℃上清、28℃沉淀,加 iptg 37℃上清、37℃沉淀分别点样跑蛋白胶,观察重组蛋白sod表达水平,以确定其在沉淀还是上清中表达,来判断蛋白的可溶性;sod重组蛋白的纯化:应用亲和层析法纯化带组氨酸标签的sod重组蛋白,跑蛋白胶,观察重组蛋白sod大小,以确定其是否纯化出sod重组蛋白;根据蛋白大小公式计算,sod基因蛋白大小为26.4kda,而pet
‑
32a原核表达载体含的his(组氨酸标签)蛋白大小约为21kda,所以pet
‑
32a
‑
sod蛋白大小约为47.4 kda;当诱导剂iptg含量为1mmol/l,诱导温度为28℃,诱导时间为6h或8 h时蛋白诱导表达效果最佳(图2)。可溶性蛋白检测。加入1mmol/l 的 iptg 诱导剂28℃或37℃诱导6h,上清和沉淀用 sds
‑
page 凝胶电泳分析,由图可知当诱导剂 iptg含量为1mmol/l,诱导温度为28℃,诱导时间为6h时,上清诱导表达蛋白量多于沉淀诱导表达蛋白量,上清多为可溶性蛋白,沉淀多为包涵体(图3);说明 pet
‑
32a
‑
sod 重组蛋白大部分在上清中表达,sod原核表达蛋白为可溶性蛋白;应用亲和层析法纯化带his标签的sod原核表达重组蛋白;向od
600
=0.6
‑
0.8的600ml的菌液中加入1mmol/l的iptg,28℃摇床诱导培养6h,破碎,离心,取上清过his标签的层析柱进行蛋白纯化,梯度洗脱纯化的重组蛋白,sds
‑
page凝胶电泳染色脱色观察(图4),经过去除杂蛋白和梯度洗脱后,加入100%的洗涤缓冲液b洗脱得到的洗脱液47.4 kda,基本为纯蛋白,说明sod原核表达重组蛋白纯化成功。
9.3、将天麻sod基因的cdna片段连接至植物过量表达载ph2gw7中,该载体含有增强型启动子,可在受体植物中过量表达目的基因;利用根癌农杆菌介导法,将目的基因转入到受体蜜环菌中,利用pda培养基培养的方法获得过量表达超氧化物歧化酶基因的转基因型蜜环菌,并通过进一步实验验证该基因是否具有提高蜜环菌抗寒能力的特性;结果表明过量表达该超氧化物歧化酶基因的蜜环菌相对于野生型蜜环菌具有更强的抗寒特性;4、利用根癌农杆菌介导法,将目的基因转入到受体拟南芥中,结果表明过表达该超氧化物歧化酶基因的拟南芥生长较快。
10.本发明的优点和技术效果:本发明所提供的基因应用了转基因的方法,改变逆境下蜜环菌关键基因的表达,
有助于缩短蜜环菌的培养时间,提高工程菌抗低温胁迫作用,同时该发明为扩大天麻种植范围、提高栽培天麻的产量和质量、天麻抗寒性、促进天麻生长发育以及提高抗寒天麻的育种提供理论基础,同时天麻超氧化物歧化酶sod基因能促进拟南芥生长,为超氧化物歧化酶基因促进植物生长研究提供理论基础。
附图说明
11.图1为pmd
‑
18t
‑
sod(a)、pet
‑
32a空载(b)和pet
‑
32a
‑
sod(c)质粒双酶切及bl21菌液pcr(d)检测电泳图,a图中m:dl15000bpdnamarker,1
‑
5:pmd
‑
18t
‑
sod质粒双酶切;b图中m:dl5000bpdnamarker,1
‑
2:pet
‑
32a空载质粒双酶切;c图中m:dl5000bpdnamarker,1
‑
4:pet
‑
32a
‑
sod质粒双酶切检测;d图中m:dl5000bpdnamarker,1
‑
6:pet
‑
32a
‑
sod转入bl21菌液pcr产物检测;图2为28℃下不同时间诱导sod原核蛋白表达结果,其中m为116kda蛋白marker(blueplus@iiproteinmarker),ck:没加iptg,其余为分别加1mol/l的iptg诱导0、2、4、6、8h;图3为sod重组表达蛋白的可溶性分析;图中m120kda蛋白marker(blueplus
@
iiproteinmarker),1:不加iptg沉淀,2:不加iptg上清;3:37℃6h沉淀,4:37℃6h上清;5:28℃6h沉淀,6:28℃6h诱导上清;图4为是sod基因重组蛋白的纯化结果,其中m:120kda蛋白marker(blueplus@iiproteinmarker),裂解液:sod原核表达全菌裂解液,其余为洗脱液;图5为sod基因未知片段扩增(a)和sod未知片段菌液pcr(b)电泳检测,a图中m:dl2000bpdnamarker,1
‑
9:sod基因未知片段扩增pcr产物(250bp)检测;b图中m:dl2000bpdnamarker,1
‑
8:sod基因未知片段菌液pcr产物(250bp)检测;图6为sod基因全长扩增(a)及pmd
‑
18t
‑
sod菌液(b)pcr检测电泳图;a图中m:dl2000bpdnamarker,1
‑
10:sod基因全长扩增pcr产物(720bp)检测;b图中m:dl2000bpdnamarker,1
‑
11:pmd
‑
18t
‑
sod菌液pcr产物(720bp)检测,12:水作空白对照;图7为pmd
‑
18t
‑
sod(a)和pentr2b
‑
sod(b)质粒双酶切检测电泳图;a图中m:dl15000bpdnamarker,1
‑
5:pmd
‑
18t
‑
sod质粒双酶切;b图中m:dl5000bpdnamarker,1
‑
5:pentr2b
‑
sod质粒双酶切检测;图8为过表达载体ph2gw7.0
‑
35s
‑
sod转化农杆菌pcr检测电泳图;其中m:dl2000bpdnamarker,1
‑
6:转化的农杆菌;7为水空白对照;图9为sod过表达转基因蜜环菌的生长情况结果示意图,a图是阳性克隆转基因蜜环菌,b图是sod基因相对表达量比较结果,c图是野生型am02,d图是过表达ph2gw7.0
‑
35s
‑
sod蜜环菌;图10为t
‑
dna纯合突变体拟南芥的筛选结果,图中m为dl2000bpdnamarker,1
‑
2是野生型拟南芥;3、4是水空白对照;5
‑
11是纯合突变体;图11为筛选转化成功的拟南芥的结果,图中水为空白对照,col
‑
0为野生型拟南芥,11c为salk_015511c突变体,sod为sod基因转化成功的拟南芥,其余泳道为未转化成功的拟南芥;
图12为sod过表达转基因拟南芥的生长情况结果示意图,a图纯合sod突变体拟南芥;b图为野生型col
‑
0拟南芥,c
‑
f图为sod过表达转基因拟南芥。
具体实施方式
12.下面通过实例和附图对本发明作进一步详细说明,但本发明保护范围不局限于所述内容。实施例中方法如无特殊说明,按常规操作进行,如无特殊说明使用试剂均为常规购试剂或按常规方法配制的试剂,如无特殊说明方法中百分数均为质量百分数。
13.实施例1:本发明天麻超氧化物歧化酶(superoxide dismutase,sod)基因的获取选取13℃下培养的母麻为实验材料,采用trizol reagent(invitrogen)法提取天麻的总rna,具体是用研钵将0.15g 13℃母麻样品研磨成粉末,加入1ml的trizol提取液在研钵中继续研磨至液体透明,室温静置5min后移入离心管,再加入0.2 ml氯仿,振荡混匀,4℃、12000rpm离心15min,转移上清液至新管,重复加入200μl氯仿一次取上清,加入与上清液等体积的异丙醇200μl 和柠檬酸钠高盐溶液200μl(用于去除天麻中的多糖),混匀
‑
20℃静置30min,4℃、12000rpm离心30min,弃上清,沉淀用75%乙醇1ml清洗3次,4℃、7500rpm离心5min,弃乙醇真空干燥沉淀或自然晾干,用20μl稀释1000倍的焦碳酸二乙酯(depc)溶解rna,放
‑
80℃保存备用。
14.用prime script rt reagengt rit with gdna eraser试剂盒反转总rna形成cdna,具体步骤如下:(1)基因组dna去除,8μl rnase free ddh2o、4μl 4
×
gdna wiper mix、1μl oligo(dt)23 vn(50μm)、1μl random hexamers(50ng/μl)和2μl总 rna混匀,42℃加热2min;(2)配制第一链 cdna 合成体系,在步骤(1)中加入2μl 10
×
rt mix和2μl hiscript ii enzyme mix混匀,50℃下加热15min后,85℃加热2min,得反应产物 cdna,置于
‑
20℃或
‑
80℃备用。
15.从转录组数据中筛选得到的sod基因的cds是只有5’端的碱基序列,而3’端部分碱基未知,通过dnaman和primer premier5软件设计sod基因的巢式pcr引物(sod
‑
f1:5
’‑
gaacatgcgtactaccttcag
‑3’
;sod
‑
f2:5
’‑
gcgaggtgtacgaggctgag
‑3’
;un36:5
’‑
gactcgagtcgacatcgatttttttttttttttttt
‑3’
)和全长扩增的引物sod
‑
f:5
’‑
ggtaccatggcgctccgagccac
‑3’ꢀ
(上游:kpni);sod
‑
r:5
’‑
gatatcttattccaccacctcagc
‑3’ꢀ
(下游:ecor v),全长扩增添加了酶切位点序列,以便构载体时进行双酶切。sod 基因序列上游为 kpn i 核苷酸内切酶(酶切位点序列为 ggtacc),下游为ecorv 核苷酸内切酶(酶切位点序列为gatatc);引物由昆明硕擎公司进行合成。
16.进行两次巢式pcr,第一次巢式 pcr:(1)扩增体系:0.7 μl上游引物(sod
‑
f1)和 0.7 μl 下游通用引物(un36),2 μl天麻的cdna 模板,10 μl的 2
×
es taq master mix,6.6μl ddh2o。(2)反应条件:预变性 94℃ 3 min;(变性 94℃ 40s,退火 53℃ 30s,延伸 72℃ 2min)30个循环;再延伸 72℃ 10min。 第二次巢式 pcr:(1)扩增体系:0.7 μl 上游引物(sod
‑
f2)和 0.7 μl下游通用引物(un36),2 μl第一次巢式 pcr 产物稀释 10 倍的 dna 模板,10 μl的2
×ꢀ
es taq master mix,6.6 μl ddh2o。(2)反应条件:退火 60℃ 30s。
17.将第二次巢式pcr的反应产物进行跑胶检测,将检测到的目的未知片段进行胶回收,使用sanprep柱式dna胶回收试剂盒(上海生工购买)进行胶回收,具体步骤如下:
①
切下
目的基因凝胶称量,加入凝胶重量 4倍的buffer b2,50℃加热至完全融化;
②
移入吸附柱 8000g 30s,弃收集管液体,可重复此操作;
③
加入500μl wash solution 9000g离心30s,弃收集管液体,重复一次此操作,将空吸附柱 9000 g离心1min;
④
加入30μl提前60℃预热的 elution buffer,室温静置 1
‑
2min 后9000g离心1min;
⑤
将回收的目的未知片段 dna 跑胶检测 dna 回收情况,结果见图5a,sod基因未知片段的电泳图,可知未知片段在250bp以内,且9个重复条带一样大;使用pmd
‑
18t vector kit(takara公司购买)进行ta克隆,反应体系如下:pmd
‑
18t 载体1μl,solution i 5 μl,目的未知片段 dna 4μl,混匀后置于 16℃反应4h或过夜;采用热击法转化感受态细胞dh5α大肠杆菌(购买于上海擎科生物有限公司),具体步骤如下:将10μl ta克隆的反应体系,转入到不完全融化的100μl 感受态细胞dh5α 中,混匀,置于冰上30min,42℃加热45s,再置于冰上3min,加入890μl 无抗生素 lb 液体培养基,于37℃、180r/min 培养1h 后,5000rpm离心2min,将沉淀涂于添加100μg/ml amp 抗生素的 lb 固体培养基中,37℃培养12 h,挑单菌落于10ml 相同抗性液体lb培养基中,摇床培养至 od600=0.6。用sod
‑
f2、un36 引物进行pcr检测,结果见图5b,条带也是在250bp左右;所以初步判定sod基因的3’端序列,在250bp以内;未知片段菌液pcr阳性克隆送上海擎科生物有限公司进行测序,将获得的测序结果与引物序列(sod
‑
f2、un36)进行比对,并将比对成功的序列与sod基因已知的片段序列进行拼接,使用dnaman 软件查找终止密码子,得到sod 基因全长cds序列(720bp),核苷酸序列如seq id no:1所示,氨基酸序列如seq id no:2所示,编码239个氨基酸残基;用天麻 cdna 作为模板,sod
‑
f和sod
‑
r作为引物,pcr 扩增sod基因全长序列,pcr 扩增全长具体步骤为:(1)扩增体系:0.5μl上游引物(sod
‑
f)、0.5μl下游引物(sod
‑
r)、1μl天麻的cdna模板、10μl 的2
×
es taq master mix、8 μl ddh2o。(2)反应条件:预变性 94℃ 3 min,(变性 94℃ 40 s,退火58℃ 30 s,延伸 72℃ 2 min)32 个循环,再延伸 72℃ 10 min。pcr 产物跑胶,将目的片段进行胶回收(参照上述方法)结果见图6a得到一条约750bp左右的序列,进行ta 克隆(参照上述方法),pmd
‑
18t
‑
sod菌液 pcr得到也一条750bp左右的序列(图6b),筛选阳性克隆进行双向测序,通过 dnaman 比对,提取质粒送测得到的测序结果与拼接好的序列相似率为100%,说明pmd
‑
18t
‑
sod 连接成功。
18.实施例2:sod基因真核表达载体的构建采用sanprep柱式质粒dna小量抽提试剂盒(上海生工)提取的pmd
‑
18t
‑
sod质粒和pentrtm
‑
2b质粒分别进行ecor v和kpn i双酶切(20μl体系),反应体系和操作过程为:取2μl pmd
‑
18t
‑
sod或pentrtm
‑
2b质粒、依次加入2μl 10
×
k buffer、0.5μl kpn i、0.5μl ecor v、15μl ddh2o,混匀后置于37℃反应3h;分别进行胶回收。
19.将回收的sod目的基因片段和pentrtm
‑
2b载体片段进行连接,转化 dh5α 感受态细胞,涂于含100
µ
g/ml kan抗性的lb固体上37℃培养12h,挑取单菌落于20ml 相同抗性lb 液体培养基中37℃培养12h,提取质粒双酶切检验,送测比对是否连接上入门载体。
20.将检测正确的入门克隆载体pentrtm
‑
2b
‑
sod与目的getway载体ph2gw7.0进行lr反应,采用gateway lr clonase tm ii enzyme mix试剂盒,具体步骤如下:
①
配制体系:6
µ
l 入门载体 pentrtm
‑
2b
‑
sod 质粒,3
µ
l目的载体 ph2gw7.0 质粒,1
µ
l 从
‑
80℃取出漩涡震荡 2 次后的 lr clonase tm ii enzyme mix,混匀;
②
lr反应:将体系置于25℃反应4h
或过夜后,在体系中加入1
µ
l proteinase k,37℃反应10min 终止lr反应。转化dh5α,涂布于50
µ
g/ml spe抗性的lb固体上37℃培养12h,挑取单菌落于相同抗性的lb液体培养基中37℃摇床培养12h,提取ph7wg2.0
‑
35s
‑
sod质粒、双酶切检测,送质粒检测,对比过表达ph7wg2.0
‑
35s
‑
sod载体是否构建成功。
21.将构建成功的过表达载体质粒ph2gw7.0
‑
35s
‑
sod用电击法转化入农杆菌pmp90中,通过电转化法将ph7wg2.0
‑
35s
‑
sod转入农杆菌感受态pmp90中。具体步骤如下:
①
将电转杯用75%酒精洗净,无菌下风干,
‑
20℃预冷2min;
②
取2
µ
lph7wg2.0
‑
35s
‑
sod质粒于pmp90农杆菌感受态细胞中,混匀加入到预冷的电转杯中;
③
将电转杯放置电转化槽中进行电转化;
④
电击后,立即取出电转杯,将菌液加入到900
µ
l的无抗性lb液体培养基中,28℃孵育4h,7500rpm离心2min,菌体涂于100
µ
g/ml spe抗性的lb固体平板上28℃培养24~48h,挑取单菌落于相同抗性的lb液体培养基中28℃培养24~48 h,pcr 鉴定单克隆农杆菌菌株。
22.结果构建过表达载体的入门载体pentr2b
‑
sod。pmd
‑
18t
‑
sod质粒酶切检验(图7a)切开得到目的条带大小为720bp的条带后送测,用正确pmd
‑
18t
‑
sod质粒和pentr2b空载质粒进行双酶切后将目的基因片段和入门载体片段胶回收,用solution i将其连接,构成pentr2b
‑
sod入门载体,经过kpnⅰ和ecor v双酶切检验(图7b)正确后送测序,比对成功后;说明入门载体pentr2b
‑
sod构建成功。将构建成功的gateway入门载体pentr2b
‑
sod使用gateway lr clonase tm
ꢀⅱ
enzyme mix试剂盒,进行lr反应,转化 dh5α菌液pcr和双酶切检测构建情况,阳性质粒送测,得到过表达载体ph2gw7.0
‑
35s
‑
sod。过表达载体ph2gw7.0
‑
35s
‑
sod用电击法转化农杆菌pmp90菌液pcr检测转化情况(图8),阳性质粒送测,表明ph2gw7.0
‑
35s
‑
sod农杆菌转化成功,可用于后续转化蜜环菌菌丝。
23.实施例3:过表达sod真核载体转化蜜环菌am02农杆菌转染蜜环菌am02形成基因工程蜜环菌:
①
将阳性克隆农杆菌在30ml 100
µ
g/ml spe抗性培养基中进行扩培至od600=1.0左右,4℃、3000rpm离心10min,将沉淀重悬于5ml含150
µ
mol/l乙酰丁香酮(as)的诱导培养基中,28℃摇床培养至od600=1.2。
②
将蜜环菌菌球用超声匀浆机破碎混匀,避光4℃静置培养3h。
③
将诱导农杆菌按1:1的比例加入到避光菌丝体中混匀,25℃共培养10h,离心去培养基,使用含400
µ
g/ml cef(头孢噻肟钠)抗生素无菌纯净水洗涤多次后,涂布于含100
µ
g/ml spe抗性的pda固体培养基上,25℃避光培养至蜜环菌长出。
24.sod基因工程蜜环菌验证:挑取转基因蜜环菌于含100
ꢀµ
g/ml spe抗性的pda培养基中培养,采用trizol reagent(invitrogen)法提取ph2gw7.0
‑
35s
‑
sod密环菌菌索rna和野生型实验室分离的2号蜜环菌(wt
‑
am02)菌索的rna;用prime script rt reagengt rit with gdna eraser试剂盒反转总rna形成cdna;使用primer premier 5.0软件设计引物:sod基因引物(f
‑
tcaccaccagaagcaccatcaa;r
‑
ggacaaccttggaagcatccct)和内参基因β
‑
actin(f
‑
ggggatgaagcacagtccaa ;r
‑
gccgtggttgtgaaggagta),做q
‑
pcr检验,用2
‑
δδct方法计算基因表达水平。用q
‑
pcr方法对每个样品的至少两个独立的生物复制和三个技术复制进行了分析,以确重复性和可靠性。
25.将构建的过表达载体 ph2gw7.0
‑
35s
‑
sod 通过电转法转化农杆菌,25℃下在诱导培养基中与野生型蜜环菌am02共培养后,将蜜环菌菌丝体用水和加 cef 抗生素的水清洗至液体清澈,涂于含hyg抗性的pda固体培养基上生长至1个月筛选阳性克隆转基因蜜环菌,
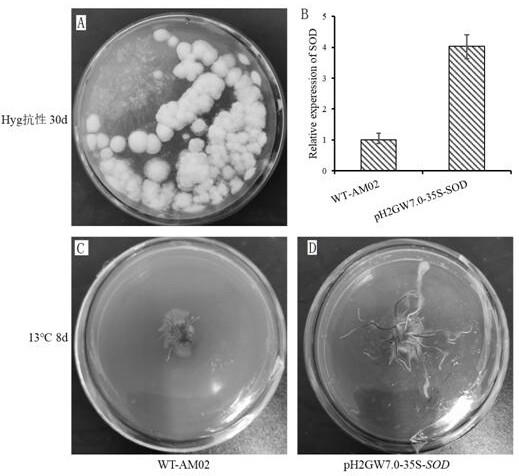
发现长出(图9a)的蜜环菌菌落,将其挑于新的相同抗性pda上继续培养,实验表明在hyg抗性pda上正常生长,说明sod转化蜜环菌成功。
26.将转sod基因工程蜜环菌和野生型蜜环菌am02分别制备成菌索后,取同样大小的菌索在pda培养基于13℃下培养,8d后13℃过表达ph2gw7.0
‑
35s
‑
sod蜜环菌菌索(图9d)长势比野生型am02(图9c)长势好且快速,说明转入sod基因的蜜环菌有更好的抗低温逆境胁迫作用;还通过q
‑
pcr分析发现13℃培养的ph2gw7.0
‑
35s
‑
sod密环菌菌索中sod基因相对表达量高于野生型am02(图9b),说明sod转基因工程蜜环菌转化成功,sod 基因有助于缩短蜜环菌的培养时间和提高蜜环菌的抗寒性,进一步有助于缩短天麻生产的周期。
27.实施例3:天麻超氧化物歧化酶基因在促进拟南芥生长实验1、拟南芥种质(germplasm)为salk_015511c(superoxide dismutase)突变体种子,购于abrc(https://abrc.osu.edu/);拟南芥种植:野生型和突变体拟南芥种子表面消毒;将消毒后的拟南芥种子播种于ms培养基上,4℃黑暗春化2d,然后置于23℃,光照强度为60
‑
100
µ
mol/m
2.
s,连续光照下培养,约7d开始发芽,待长20d左右长出根和3
‑
4片真叶后,移栽至土壤培养1个月左右,用于鉴定纯合拟南芥突变体。
28.根据拟南芥信息数据库查找t
‑
dna设计lp、rp、lb三引物,在http//:signal.edu/ tdnaprimers.2.html查询lp和rp引物,在www.arabidopsis.org /index.isp查找适用于该种质的lb引物;sod11c
‑
lp:5
’‑
tcaatgcatcattttgctttg
‑3’
;sod11c
‑
rp:5
’‑
aatccagctgtgataacaccg
‑3’
;lb_6313r
‑
lb:5
’‑
tcaaacaggattttcgcctgct
‑3’
;拟南芥纯合sod突变体筛选:用一步法提取拟南芥dna,取0.15g拟南芥叶片加液氮研磨成粉末,加入200
µ
l dna提取缓冲液[edwards 缓冲液:200mmol/l tris
‑
hcl(ph7.5),250mmol/l nacl,25mmol/l edta,0.5%sds和te缓冲液:10mmol/l tris
‑
hcl(ph8.0),1mmol/l edta,用te缓冲液将edwards 缓冲液稀释10倍,现配现用]混匀,4℃、14000rpm离心5min,得上清,将含有拟南芥dna的上清电泳检测。pcr检验纯合突变体,以野生型(wt)作为对照,采用t
‑
dna的特异引物lp和rp各0.5
µ
l进行pcr,以salk_015511c突变型作为实验组,用三引物sod11c
‑
lp、sod11c
‑
rp、lb_6313r
‑
lb各0.5
µ
l进行pcr,pcr条件为94℃预变性3min,94℃变性30s,55℃退火30s,72℃延伸40s,30 个循环,72℃再延伸10min。纯合突变跑胶验证:电泳检测纯合情况,根据条带大小及条数分辨出突变体纯合子与杂合子;种质salk_015511c筛选见(图10),5
‑
11泳道对应的植株为纯合突变体;与野生型对比,纯合突变体只有一条带,且小于野生型(rp bp);若为杂合则应有两条带,一条与野生型大小相同(rp lp),一条小于野生型(rp bp);2、f1代拟南芥纯合突变体栽培:收集筛选出的纯合突变体植株的种子,消毒点种于ms固体培养基上培养,然后移栽至土壤中,选取生长茁壮的,适时打顶,使其长出更多的侧枝花序;3、冻融法转化农杆菌:取2
µ
l ph7wg2.0
‑
35s
‑
sod质粒加入到100
µ
l
ꢀ‑
80℃保存得pmp90农杆菌感受态细胞中,混匀冰上孵育30min,液氮冻5min,37℃ 5min,冰上2min,加入890
µ
l无抗性lb液体培养基,28℃摇床恢复培养4h,涂于100
µ
g/ml spe抗性的lb固体平板
上,28℃培养24
‑
48h,挑取单菌落于相同抗性的lb液体培养基中,28℃摇床培养24
‑
48h,pcr鉴定单克隆农杆菌菌株。
[0029]
4、摇菌:选取pcr验证成功的菌液,按1%接种量接种于150ml 100
µ
g/ml spe抗性的lb液体培养基中,28℃培养至od600=1.0左右,5000rpm离心10min,菌体沉淀加入菌体等体积的5%蔗糖溶液重悬后,加入0.1%的silwet l
‑
77表面活性剂,摇匀;5、侵染:将待侵染纯合sod突变体f1代拟南芥提前一天交足水,剪去果荚和完全开放的花序,将花序侵染于步骤4中制备的菌液中静置60s,真空干燥1min,重复操作1次,用保鲜膜包好,横着黑暗放置18h揭下保鲜膜,避免超过20h,会大大增加花序的焉萎,正着放置生长即可;侵染7d后可进行第二次侵染,不能剪果荚和花序;待种子成熟,收种子标记好,室温保存;6、筛选sod阳性克隆拟南芥植株:侵染种子点种于spe抗性ms培养基中培养1个月后,取0.15g拟南芥叶片用一步法提取拟南芥dna,用引物sod
‑
f和sod
‑
r筛选转化成功的拟南芥植株,结果见图11;7、将转化成功的拟南芥的种子点种于ms培养基中培养15天后,拟南芥生长情况见图12,图中显示sod过表达转基因拟南芥(图12c
‑
f)比野生型拟南芥(图12b)、拟南芥纯合sod突变体(图12a)长势都好;结果表明说明天麻sod基因能促进拟南芥生长。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

