本发明属于磷光材料技术领域,涉及一种室温磷光材料及其制备方法及应用。
背景技术:
室温磷光材料由于其具有较长的发射寿命(毫秒到秒级),较大的stokes位移和丰富的激发态能级结构,在光电器件制备、化学传感和生物成像领域具有较高应用价值。
目前,基于有机金属络合物和纯有机化合物的室温磷光材料得到了广泛研究。然而,有机金属配合物通常含有贵金属,成本高且具有潜在生物毒性(j.am.chem.soc.2015,137,5304;j.am.chem.soc.2013,135,14125.);另一方面,纯有机化合物通常需要引入特定的基质与晶体结构,导致其水分散性较差(nat.mater.2015,14,685;sci.adv.2018,4:eaas9732.)。
因此,亟需一种具有良好水溶性和较低生物毒性的室温磷光材料。
技术实现要素:
针对上述不足,有必要提供一种新的室温磷光材料。
一种室温磷光材料,所述室温磷光材料为纳米球状粉末,所述纳米球状粉末包括氧化硅骨架、以及掺杂在所述氧化硅骨架中的锌离子。
上述室温磷光材料,为硅纳米材料,其具有良好的生物相容性,表面可适应性和低毒性,可广泛应用于生物和生物医学。此外,硅是人体中常见的微量成分,众多组织中天然存在于多孔硅(例如原硅酸)的生物降解产物。美国食品药品监督管理局(fda)最近已将硅纳米颗粒用于人类首次临床试验(nat.mater.2009,8,331)。上述室温磷光材料,不含贵金属,避免了因贵金属引入的潜在生物毒性。上述室温磷光材料,磷光寿命约为236毫秒,荧光寿命约为13纳秒;在紫外光(365nm)的激发下可以发出蓝色荧光(发射波长440nm),关掉紫外灯,裸眼可观察到约为8秒的绿色室温磷光信号。上述室温磷光材料,具有良好的水分散性,优异的ph和光稳定性(在200分钟的紫外线照射下保持稳定的荧光和磷光发射)。上述室温磷光材料,还可以利用上述独特的光学特性,作为高信噪比的磷光探针,用于无背景生物成像。
优选地,所述纳米球状粉末的平均粒径为100~200nm。
优选地,所述锌离子的掺杂比例为0.05~0.4wt%。
本发明还提供一种上述室温磷光材料的制备方法。
一种室温磷光材料的制备方法,包括如下步骤:
将有机硅烷和还原剂分散于水中,得到分散液;
将锌源分散于所述分散液中,得到反应前体溶液;
将反应前体溶液进行水热反应;
将水热反应的固体产物分离出来,得到室温磷光材料。
上述室温磷光材料的制备方法,得到室温磷光材料,具有优异的水分散性和室温磷光性能,且生物毒性低。
优选地,所述有机硅烷选自(3-氨基丙基)三甲氧基硅烷、或(3-氨基丙基)三乙氧基硅烷;所述还原剂选自柠檬酸三钠或柠檬酸;所述锌源选自硝酸锌、氯化锌、或硫酸锌中的一种或几种。
优选地,所述水热反应的加热温度为180℃~220℃,加热时间为5h~12h。
优选地,所述将有机硅烷和还原剂分散于水中的步骤包括:
在惰性气氛下,将有机硅烷和还原剂加入到去离子水中,搅拌15min以上;
将锌源分散于所述分散液中的步骤包括:
将锌源加入到所述分散液中,搅拌15min以上;
将反应前体溶液进行水热反应的步骤包括:
将反应前体溶液转移到反应釜中,将反应釜放入烘箱中加热;
将水热反应的固体产物分离出来的步骤包括:
将水热反应完成后溶液冷却至室温,然后进行多次离心洗涤;所述离心洗涤的转速为8000转;次数为3~4次,每次5min。
优选地,以30-50毫升的水为基准,所述有机硅烷为3-5克,所述还原剂为0.2-0.5克,所述锌源中的锌元素为0.00025~0.003摩尔。
本发明还提供了一种上述室温磷光材料的应用。
一种上述室温磷光材料在生物成像中的应用。
一种上述室温磷光材料在传感分析中的应用。
附图说明
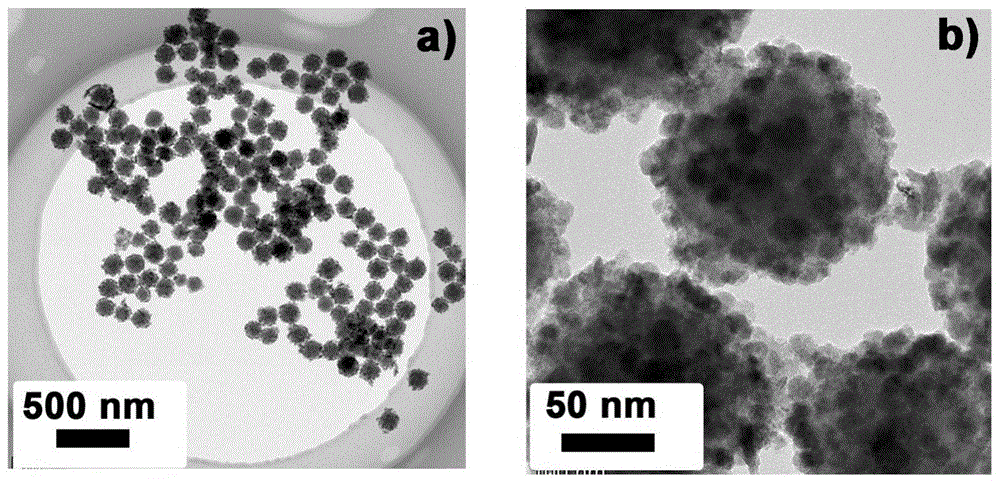
图1为实施例2的室温磷光材料的透射电镜(tem)图。
图2为实施例2的室温磷光材料的荧光和磷光特性图。
图3为实施例2的室温磷光材料的水溶液在紫外灯的辐照下及撤去紫外灯后的发光图像。
图4为实施例2的室温磷光材料荧光和磷光ph稳定性测试图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合具体实施方式,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施方式仅仅用以解释本发明,并不用于限定本发明。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本发明。本文所使用的术语“及/或”包括一个或多个相关的所列项目的任意的和所有的组合。
一种室温磷光材料,所述室温磷光材料为纳米球状粉末,所述纳米球状粉末包括氧化硅骨架、以及掺杂在所述氧化硅骨架中的锌离子。
也就是说,室温磷光材料的主体部分是氧化硅,而锌离子掺杂于主体部分中。
优选地,所述纳米球状粉末的平均粒径为100~200nm。
优选地,所述锌离子的掺杂比例为0.05~0.4wt%。
上述室温磷光材料,为硅纳米材料,其具有良好的生物相容性,表面可适应性和低毒性,可广泛应用于生物和生物医学。此外,硅是人体中常见的微量成分,众多组织中天然存在于多孔硅(例如原硅酸)的生物降解产物。美国食品药品监督管理局(fda)最近已将硅纳米颗粒用于人类首次临床试验(nat.mater.2009,8,331)。上述室温磷光材料,不含贵金属,避免了因贵金属引入的潜在生物毒性。上述室温磷光材料,磷光寿命约为236毫秒,荧光寿命约为13纳秒;在紫外光(365nm)的激发下可以发出蓝色荧光(发射波长440nm),关掉紫外灯,裸眼可观察到约为8秒的绿色室温磷光信号。上述室温磷光材料,具有良好的水分散性,优异的ph和光稳定性(在200分钟的紫外线照射下保持稳定的荧光和磷光发射)。上述室温磷光材料,还可以利用上述独特的光学特性,作为高信噪比的磷光探针,用于无背景生物成像。
本发明还提供一种上述室温磷光材料的制备方法。
一种室温磷光材料的制备方法,包括如下步骤:
s1、将有机硅烷和还原剂分散于水中,得到分散液;
s2、将锌源分散于所述分散液中,得到反应前体溶液;
s3、将反应前体溶液进行水热反应;
s4、将水热反应的固体产物分离出来,得到室温磷光材料。
其中,有机硅烷是氧化硅骨架的前驱体,锌源的目的是提供锌离子。优选地,所述有机硅烷选自(3-氨基丙基)三甲氧基硅烷、或(3-氨基丙基)三乙氧基硅烷;所述还原剂选自柠檬酸三钠或柠檬酸;所述锌源选自硝酸锌、氯化锌、或硫酸锌中的一种或几种。
在步骤s1中,优选地,所述将有机硅烷和还原剂分散于水中的步骤包括:
在惰性气氛下,将有机硅烷和还原剂加入到去离子水中,搅拌15min以上;更优选地,惰性气氛为氮气气氛。可以理解的是,惰性气氛并不局限于氮气气氛,只要是不参与反应,可以避免外界气氛造成干扰的气体均可。
在步骤s2中,锌离子作为螯合剂,锌离子与有机硅烷络合。
优选地,将锌源分散于所述分散液中的步骤包括:
将锌源加入到所述分散液中,搅拌15min以上;更优选地,搅拌过夜。
在步骤s3中,通过水热反应,有机硅烷与还原剂的高温水解以及碳化过程,使有机硅烷转变氧化硅骨架。
优选地,所述水热反应的加热温度为180℃~220℃,加热时间为5h~12h。
优选地,将反应前体溶液进行水热反应的步骤包括:
将反应前体溶液转移到反应釜中,将反应釜放入烘箱中加热。
在步骤s4中,优选地,将水热反应的固体产物分离出来的步骤包括:
将水热反应完成后溶液冷却至室温,然后进行多次离心洗涤;所述离心洗涤的转速为8000转;次数为3~4次,每次5min。
上述室温磷光材料的制备方法,得到室温磷光材料,具有优异的水分散性和室温磷光性能,且生物毒性低。
本发明还提供了一种上述室温磷光材料的应用。
一种上述室温磷光材料在生物成像中的应用。
一种上述室温磷光材料在传感分析中的应用。
上述室温磷光材料,由于其无背景信号干扰,具有广泛的生物及生物医学应用前景,可应用于高信噪比的生物成像及传感分析等领域。
以下结合具体实施例对本发明做进一步阐述。
实施例1
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.00025摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间10小时,加热温度220℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
实施例2
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.0005摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间10小时,加热温度220℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
实施例3
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.001摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间10小时,加热温度220℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
实施例4
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.003摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间10小时,加热温度220℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
实施例5
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.00025摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间8小时,加热温度200℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
实施例6
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.0005摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间8小时,加热温度200℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
实施例7
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.001摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间10小时,加热温度180℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
实施例8
将4g(3-氨基丙基)三甲氧基硅烷和0.3g柠檬酸三钠混合分散在30ml的去离子水中,持续搅拌15min以上,并通入氮气保护,得到分散液。将0.003摩尔的硝酸锌加至分散液中并搅拌过夜,得到反应前体溶液。将反应前体溶液转移至反应釜中,放入烘箱进行水热反应;反应条件为加热时间10小时,加热温度200℃。反应完成后,冷却至室温,进行离心洗涤,离心转速为8000转,每次5分钟,重复4次,得到的下层沉淀即为室温磷光材料。
性能测试
tem测试:
对实施例2的室温磷光材料作tem测试,测试结果见图1。
其中,图1(a)、(b)为不同分辨率下的tem图。从图1中可以看出,室温磷光材料呈球状纳米颗粒,且具有较好的分散性,并没有大幅团聚现象;球状纳米颗粒的粒径约为150纳米。
荧光磷光光谱以及衰减测试:
对实施例2的室温磷光材料作荧光发射光谱测试(λex=350nm),测试结果见图2(a)。
对实施例2的室温磷光材料作磷光发射光谱测试(λex=350nm),测试结果见图2(b)。
对实施例2的室温磷光材料作时间分辨的荧光衰减曲线(λex=370nm),测试结果见图2(c)。
对实施例2的室温磷光材料作磷光寿命衰减曲线(λex=350nm),测试结果见图2(d)。
从图2中可以看出,该室温磷光材料的荧光发射峰为440纳米,磷光发射峰为520纳米。通过对荧光及磷光衰减曲线进行拟合,计算得到其荧光寿命约为13纳秒,磷光寿命为236毫秒。
裸眼磷光测试:
将实施例2的室温磷光材料的粉末在紫外灯下照射,然后撤去紫外灯,裸眼观察,观察结果见图3(a)。
将实施例2的室温磷光材料的水溶液在紫外灯下照射,然后撤去紫外灯,裸眼观察,观察结果见图3(b)。
从图3(a)可以看出,实施例2的室温磷光材料的粉末在紫外灯的照射下,发出蓝色荧光;撤去紫外灯后,裸眼可观察到约8s的磷光信号。
从图3(b)可以看出,实施例2的室温磷光材料的水溶液在紫外灯的照射下,发出蓝色荧光;撤去紫外灯后,裸眼可观察到约5s的磷光信号。
稳定性测试:
对实施例2的室温磷光材料在不同的ph下作磷光和荧光强度测试,测试结果见图4(a)。
对实施例2的室温磷光材料在不同的紫外光辐射时长下作磷光和荧光强度测试,测试结果见图4(b)。
从图4(a)可以看出,不同的ph下,其磷光和荧光强度基本保持同一水平,可见ph变化对其荧光和磷光性质的并无明显影响,其ph稳定性好。
从图4(b)可以看出,200min内紫外光辐射时长下,其磷光和荧光强度依然可以保持较高水平,可见200min紫外光辐射对其荧光和磷光性质的并无明显影响,其光照稳定性好。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
本文用于企业家、创业者技术爱好者查询,结果仅供参考。