一种靶向fap的双氧水响应的前药及其制备方法与应用
技术领域
1.本发明属于前药制备技术领域,尤其是涉及一种靶向fap的双氧水响应的前药及其制备方法与应用。
背景技术:
2.目前恶性肿瘤是造成全世界人类死亡的主要原因之一,已经成为严重危害人类生命健康、制约社会经济发展的一类重大疾病。目前治疗手段主要包括传统的手术、化疗和放疗以及新兴的免疫治疗等。这些治疗方法往往都需要辅以治疗试剂,而大多治疗试剂均为小分子试剂,代谢速度快,且非特异性分布,不仅副作用大,且难以有效、特异性的富集在肿瘤部位。因此,如何将这些辅助治疗试剂精准、高效的富集在肿瘤部位仍然面临着诸多挑战。连接具有靶向单元的抗体等方法,通过抗体等的靶向性,将试剂特异性靶向递送到肿瘤区域,是提高治疗试剂在肿瘤区域富集的主要方法。但是这种方法还存在着一些问题,如抗体分子量过大,不仅给连接小分子药物带来困难,而且还有可能抑制小分子药物的功能。因此,设计开发新型的小分子药物递送系统是目前仍然需要解决的问题。
3.肿瘤相关成纤维细胞(cancer
‑
associated fibroblasts,cafs)是肿瘤微环境重要的基质细胞,和肿瘤细胞的迁移、增值以及存活息息相关。而cafs高表达一种成纤维细胞激活蛋白(fibroblast activation protein,fap),这种蛋白在正常组织不表达,是肿瘤微环境的特异性标志物。因此,靶向fap的肿瘤治疗已经引起了广泛的关注。目前已经开发了多种靶向fap的抗肿瘤试剂(专利号:201910425487.0)或者影像试剂(专利号:201880083520.x)。但是化疗药物或者成像试剂连接到靶向fap试剂时,化疗药物或者fap抑制剂的功能因为结构变化而失去了治疗功能。因此,构建新的靶向fap,且能保持独立药效的前药具有重要的意义。
技术实现要素:
4.基于现有技术中缺乏靶向fap且能保持独立药效的前药的技术现状,本发明提供一种靶向fap的双氧水响应的前药及其制备方法与应用。
5.本发明的目的可以通过以下技术方案来实现:
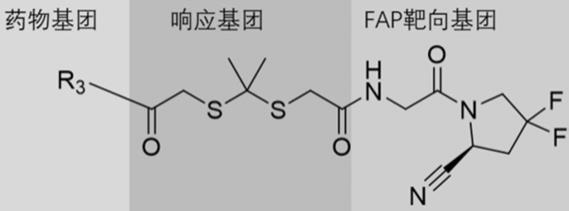
6.本发明首先提供一种靶向fap的双氧水响应的前药,其结构式如下:
[0007][0008]
其中,r3为抗肿瘤药物单元。
[0009]
所述靶向fap的双氧水响应的前药包括三部分,分别为药物基团部分,响应基团部分以及fap靶向基团部分,各部分分别示意如下所示。
[0010][0011]
其中,fap靶向基团为靶向fap的单元,具体为甘氨酰
‑2‑
氰基吡咯烷类化合物,响应基团为肿瘤内高表达双氧水响应单元,药物基团为抗肿瘤药物单元。
[0012]
在本发明的一个实施方式中,所述抗肿瘤药物选自拉罗替尼、吉非替尼、阿霉素、紫杉醇或羟基喜树碱中的一种或几种。
[0013]
本发明还提供靶向fap的双氧水响应的前药的制备方法,由带有主动靶向fap基团的物质与抗肿瘤药物通过含有双氧水响应连接基的物质,发生酰胺反应和酯化反应共价键合而得到。
[0014]
在本发明的一个实施方式中,所述带有主动靶向fap基团的物质(称之为fapi)结构如下:
[0015][0016]
上述结构的带有主动靶向fap基团的物质即为甘氨酰
‑2‑
氰基吡咯烷类化合物。
[0017]
在本发明的一个实施方式中,所述含有双氧水响应连接基的物质(称之为tk)结构如下:
[0018][0019]
所述含有双氧水响应连接基的物质中,所述连接基为含缩硫酮的小分子连接基。
[0020]
在本发明的一个实施方式中,所述含有双氧水响应连接基的物质(称之为tk)的合成方法如下:
[0021]
a、配制摩尔比为1
‑
1:2.5的丙酮与巯基乙酸的混合溶液;
[0022]
b、氮气保护下,慢慢滴加体积约0
‑
1ml的三氟乙酸,反应6
‑
36小时;
[0023]
c、待反应完成后,过滤,分别用石油醚与水洗涤得到产物,即为含有双氧水响应连接基的物质(tk)。
[0024]
在本发明的一个实施方式中,所述抗肿瘤药物(称之为r3)选自拉罗替尼、吉非替尼、阿霉素、紫杉醇或羟基喜树碱中的一种或几种。
[0025]
在本发明的一个实施方式中,靶向fap的双氧水响应的前药的制备方法包括以下步骤:
[0026]
抗肿瘤药物和含有双氧水响应连接基的物质发生反应,即首先将双氧水响应部分通过酰胺基连接到抗肿瘤药物,
[0027]
反应产物与带有主动靶向fap基团的物质发生反应,得到靶向fap的双氧水响应的前药。
[0028]
在本发明的一个实施方式中,抗肿瘤药物和含有双氧水响应连接基的物质发生反应的方法(即合成r3‑
tk)如下:
[0029]
a、配置一定浓度的2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(即hatu)的n,n
‑
二甲基甲酰胺溶液;
[0030]
b、配置tk与n,n
‑
二异丙基乙胺的二氯甲烷的溶液,其摩尔比为1
‑
1:4;
[0031]
c、将上述的b溶液缓慢加入到a溶液中,温度为0
‑
50℃,反应10
‑
30分钟;
[0032]
d、配置抗肿瘤药物(r3)和dipea的二氯甲烷中的溶液,其摩尔比为1
‑
1:4;
[0033]
e、将上述d溶液加入至上述c的溶液中,反应12
‑
36小时后,用二氯甲烷,多次萃取水相,合并有机相,有机相用无水硫酸镁干燥;
[0034]
f、过滤上述溶液,去滤液,真空除去溶剂,得粗产物,薄层层析得纯品,即r3‑
tk。
[0035]
r3‑
tk的结构如下所示:
[0036][0037]
在本发明的一个实施方式中,抗肿瘤药物和含有双氧水响应连接基的物质发生反应所得的反应产物与带有主动靶向fap基团的物质发生反应(即合成fapi
‑
r3‑
tk),方法如下,
[0038]
a、配置一定浓度的2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯的n,n
‑
二甲基甲酰胺溶液;
[0039]
b、配置r3‑
tk与n,n
‑
二异丙基乙胺的二氯甲烷的溶液,其摩尔比为1
‑
1:4;
[0040]
c、将上述的b溶液缓慢加入到a溶液中,温度为0
‑
50℃,反应10
‑
30分钟;
[0041]
d、配置fapi和dipea的二氯甲烷中的溶液,其摩尔比为1
‑
1:4;
[0042]
e、将上述d溶液加入至上述c的溶液中,反应12
‑
36小时后,用二氯甲烷,多次萃取水相,合并有机相,有机相用无水硫酸镁干燥。
[0043]
f、过滤上述溶液,去滤液,真空除去溶剂,得粗产物,薄层层析得纯品,即所述靶向fap的双氧水响应的前药。
[0044]
在本发明的一个实施方式中,靶向fap的双氧水响应的前药的合成路线如下:
[0045]
[0046]
本发明还提供所述靶向fap的双氧水响应的前药的应用,所述靶向fap的双氧水响应的前药在制备对肿瘤部位进行精准治疗的药物上的应用。
[0047]
所述靶向fap的双氧水响应的前药能够与肿瘤微环境中的双氧水反应而释放药物,对肿瘤部位进行精准治疗。
[0048]
本发明提供了靶向fap的双氧水响应的前药的制备及其应用。该前药主要包括三部分,包括靶向fap的单元(甘氨酰
‑2‑
氰基吡咯烷类化合物)、肿瘤内高表达双氧水响应单元、抗肿瘤药物单元。制备方法为:首先将双氧水响应部分通过酰胺基连接到抗肿瘤药物,随后将靶向fap的单元连接到双氧水响应单元修饰的抗肿瘤药物。
[0049]
所述靶向fap的双氧水响应的前药利用靶向fap的功能将该前药有效的富集在肿瘤部位,肿瘤内高表达的双氧水能将缩硫酮键打断,释放出抗肿瘤药物,实现抗肿瘤功能。该前药不仅能提高抗肿瘤药物在肿瘤内的富集量,且能在肿瘤内高表达的双氧水刺激下,将抗肿瘤药物释放到肿瘤内,实现抗肿瘤功能。
[0050]
与现有技术相比,本发明提供的靶向fap的双氧水响应的前药在体外无活性,进入机体后,能靶向至肿瘤区域,同时与肿瘤微环境高表达的双氧水反应而激活药物活性。
附图说明
[0051]
图1为实施例1制备得到的tk的核磁氢谱;
[0052]
图2为实施例2制备得到的ji
‑
tk的核磁氢谱;
[0053]
图3为实施例2制备得到的ji
‑
tk的核磁碳谱;
[0054]
图4为实施例3制备得到的ji
‑
tk
‑
fapi的核磁氢谱;
[0055]
图5为实施例3制备得到的ji
‑
tk
‑
fapi的核磁碳谱。
具体实施方式
[0056]
下面结合附图和具体实施例对本发明进行详细说明。
[0057]
以下各实施例中,其结构合成方法如下,若无特别说明,则表明所采用的试剂均为常规市售商品或采用本领域常规技术制备得到。
[0058]
以下实施例中靶向fap的双氧水响应的前药的合成路线如下:
[0059][0060]
实施例1 tk的合成
[0061]
配制摩尔比为1:2的丙酮(2.9g,50mmol)与巯基乙酸(11.44g,100mmol)的混合溶液,氮气保护下,慢慢滴加体积约0.1ml的三氟乙酸,反应12小时。待反应完成后,过滤,分别用石油醚、水洗涤得到产物。
[0062]
制备得到的tk的核磁氢谱如图1所示。
[0063]
实施例2 ji
‑
tk的合成
[0064]
配置2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(380mg,1mmol)的n,n
‑
二甲基甲酰胺(2ml)溶液,得溶液a。配置tk(224mg,1mmol)与n,n
‑
二异丙基乙胺(0.2ml)的10ml二氯甲烷溶液,得溶液b。将上述的b溶液缓慢加入到a溶液中,室温,反应30分钟。配置吉非替尼(446mg,1mmol)和n,n
‑
二异丙基乙胺(0.4ml)的二氯甲烷中的溶液,得溶液c。将上述c溶液加入至上述a、b的混合溶液中,反应12小时后,用二氯甲烷,多次萃取水相,合并有机相,有机相用无水硫酸镁干燥。过滤上述溶液,去滤液,真空除去溶剂,得粗产物,薄层层析得纯品,即ji
‑
tk。
[0065]
制备得到的ji
‑
tk的核磁氢谱如图2所示,核磁碳谱如图3所示。
[0066]
实施例3 ji
‑
tk
‑
fapi的合成
[0067]
配置2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(380mg,1mmol)的n,n
‑
二甲基甲酰胺(2ml)溶液,得溶液a。配置ji
‑
tk(224mg,1mmol)与n,n
‑
二异丙基乙胺(0.2ml)的10ml二氯甲烷溶液,得溶液b。将上述的b溶液缓慢加入到a溶液中,室温,反应30分钟。配置fapi(446mg,1mmol)和n,n
‑
二异丙基乙胺(0.4ml)的二氯甲烷中的溶液,得溶液c。将上述c溶液加入至上述a、b的混合溶液中,反应12小时后,用二氯甲烷,多次萃取水相,合并有机相,有机相用无水硫酸镁干燥。过滤上述溶液,去滤液,真空除去溶剂,得粗产物,薄层层析得纯品,即ji
‑
tk
‑
fapi。
[0068]
制备得到的ji
‑
tk
‑
fapi的核磁氢谱如图4所述,核磁碳谱如图5所示。
[0069]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。
熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

