一种ncas3单链核酸内切酶及其应用
技术领域
1.本发明属于生物技术领域,尤其涉及一种ncas3单链核酸内切酶及其应用。
背景技术:
2.crispr
‑
cas是一种广泛存在于大多数细菌和古菌中的原核适应性免疫系统,用于防止病毒或质粒等外源性遗传物质的入侵。
3.近年来的研究证实,crispr
‑
cas系统在结构上由两部分组成。首先,是有规律成簇间隔短回文重复序列(crisprs),宿主可以在其中存储入侵的遗传物质信息。其次,是一些与crispr相关的蛋白质(cas蛋白)。cas蛋白包括由crispr相关基因编码的解旋酶、核酸酶和结构蛋白。当储存在crispr簇中的外源基因序列被转录加工成具有特定结构的pre
‑
crrna后,cas蛋白会与crrna结合,识别并切割入侵的外源基因序列。
4.目前,原核生物适应性免疫系统已被广泛应用于微生物学、分子生物学等多个领域。迄今为止,crispr
‑
cas系统的分类层次大体上总共分为两个大类和六个型。其中的1类系统,包括i型、iii型和iv型,其特征是由多个cas蛋白共同构成。type i和type iii是最常见和多样化的,在大量的古细菌中都有存在,在细菌中则较少出现。iv型系统缺乏crispr
‑
cas基因座的适应模块。第2类系统,包括ii型、v型和vi型,这类系统则是由一个单一的、大的、多结构域的cas蛋白组成。每一种类型的crispr
‑
cas系统中都存在与之对应的特征蛋白。例如ii型系统中具有核酸内切酶活性的效应蛋白cas9和v型系统中的效应蛋白cpf1。而在ⅰ型系统中,特征效应蛋白则为cas3蛋白。
5.cas3蛋白是由cas3基因编码的一种多功能蛋白,同时具有单链dna(ssdna)刺激的atp酶活性、atp依赖的解旋酶活性和一种金属依赖的单链核酸酶活性。大多数情况下,其解旋酶结构域会与核酸酶结构域中位于cas3蛋白的n端的hd位点融合,并参与目标dna的裂解。
技术实现要素:
6.针对现有技术存在的问题,本发明提供了一种ncas3单链核酸内切酶及其应用,目的在于解决现有技术中的一部分问题或至少缓解现有技术中的一部分问题。
7.本发明是这样实现的,一种ncas3单链核酸内切酶,所述ncas3单链核酸内切酶为在野生型cas3蛋白的解旋酶功能域上引发突变,使野生型cas3蛋白丧失解旋酶活性,只具备单链核酸酶活性的ncas3蛋白。
8.进一步地,所述野生型cas3蛋白的氨基酸序列如seq id no.1所示;
9.所述ncas3单链核酸内切酶为k458a、或d608a、或r887a;
10.所述k458a的氨基酸序列与seq id no.1相比,区别仅在于将第458位的“k”替换为“a”;
11.所述d608a的氨基酸序列与seq id no.1相比,区别仅在于将第608位的“d”替换为“a”;
12.所述r887a的氨基酸序列与seq id no.1相比,区别仅在于将第887位的“r”替换为“a”。
13.进一步地,所述ncas3单链核酸内切酶的氨基酸序列为由k458a、或d608a、或r887a的氨基酸序列经过若干个氨基酸残基的取代和/或缺失和/或插入,且具有相同蛋白功能的氨基酸序列。
14.进一步地,所述野生型cas3蛋白的核苷酸序列如seq id no.2所示;
15.所述k458a的核苷酸序列与seq id no.2相比,区别仅在于将第1372
‑
1373位的“aa”替换为“gc”;
16.所述d608a的核苷酸序列与seq id no.2相比,区别仅在于将第1823位的“a”替换为“c”;
17.所述r887a的核苷酸序列与seq id no.2相比,区别仅在于将第2659
‑
2660位的“cg”替换为“gc”。
18.进一步地,所述ncas3单链核酸内切酶的核苷酸序列为与k458a、或d608a、或r887a的dna序列杂交且编码相同功能蛋白质的dna序列;或与k458a、或d608a、或r887a限定的dna序列具有90%以上同源性,且编码相同功能蛋白质的dna分子。
19.本发明还提供了一种转化体,包含如上述的ncas3单链核酸内切酶的核苷酸序列。
20.进一步地,所述转化体包括重组载体、表达盒、转基因细胞、重组菌中的任一种。
21.进一步地,重组载体可以为pet28a;重组菌可以是大肠杆菌,如bl21(de3),可以是运动发酵单胞菌。
22.进一步地,所述重组载体包含至少一个启动子。
23.进一步地,所述启动子包括增强型启动子和/或组成型启动子,它们可单独使用或与其它的启动子结合使用。
24.进一步地,重组载体还包括增强子。
25.进一步地,所述增强子包括翻译增强子或转录增强子。这些增强子区域可以是atg起始密码子或邻接区域起始密码子等,但必需与编码序列的阅读框相同,以保证整个序列的正确翻译。所述翻译控制信号和起始密码子的来源是广泛的,可以是天然的,也可以是合成的。翻译起始区域可以来自转录起始区域或结构基因。
26.本发明还提供了如上述的一种ncas3单链核酸内切酶在切割dna单链和/或基因编辑中的应用。
27.进一步地,所述基因编辑包括基因敲除、定点突变、插入中的任一种。
28.本发明还提供一种生产ncas3单链核酸内切酶的方法,该方法包括培养上述的转化体并由培养产物中收集ncas3单链核酸内切酶。收集的ncas3单链核酸内切酶可以进一步进行纯化。
29.本发明还提供一种引物对,用于扩增上述的外切
‑
α
‑
1,4
‑
糖苷酶的编码基因全长及其任意片段。引物对的序列如seq id no.3和seq id no.4所示。
30.上述任一所述蛋白质、上述任一所述编码基因、上述任一所述重组表达载体、所述表达盒、转基因细胞系或重组菌中的任意一种在内切单链dna核酸的应用也属于本发明的保护范围。
31.本发明还提供了一种运动发酵单胞菌zymomonas mobilis zm4(seo et al.2005)
基因组改造后的菌株drm2。该菌株是在原本zm4菌株的基础上,将编码cas3蛋白的zmo0681基因内的676,661号的腺嘌呤核苷酸原位替换为胞嘧啶核苷酸得到的。
32.本发明还提供了改造后的drm2菌株的应用,利用改造后该菌株内源的i
‑
f型crispr
‑
cas系统进行基因组大片段敲除时,相较于原始菌株而言,可以在基因的靶向位点处使dna发生单链断裂,并可以显著得提高编辑质粒的转化效率以及编辑效率。
33.为了达到以上目的,本发明采取了以下措施:
34.通过比对zm4菌株与其他同样携带内源i型crispr
‑
cas系统的菌株,内源crispr相关蛋白的编码基因序列差异,找到了其cas3蛋白的功能结构划分。并且通过探究i
‑
f型crispr
‑
cas系统的工作原理,设计并替换了zm4菌株内cas3蛋白解旋酶结构域中的一个氨基酸。将原本zm4菌株上编码cas3蛋白的zmo0681基因内的676,661号的腺嘌呤核苷酸原位替换为胞嘧啶核苷酸,使cas3蛋白丧失解旋酶活性,转变为只携带单链dna核酸内切酶活性的ncas3蛋白。从而得到改造后菌株drm2。
35.drm2菌株与野生型zymomonas mobilis zm4菌株的生长条件及生长曲线相同。
36.drm2菌株在完成针对基因组上大片段基因敲除实验过程中,相较于野生型zm4菌株,可以在基因的靶向位点处使dna发生单链断裂,并可以显著得提高编辑质粒的转化效率以及编辑效率。
37.综上所述,本发明的优点及积极效果为:
38.1、一般情况下,在ⅰ型crispr
‑
cas系统基因编辑过程中,cas3蛋白对靶向的目的片段造成损伤的时候,首先是发挥其单链核酸酶活性,在目标区域的dna单链上切出一个缺口。然后随着cas3蛋白沿着目的dna往下游移动的同时,发挥其解旋酶活性,使得缺口附近的氢键断裂,dna双螺旋解开。于是另一条单链暴露出来,再次与cas3蛋白接触、断裂,最终形成目的dna片段的双链断裂。
39.本发明提供的ncas3单链核酸内切酶,通过破坏掉ⅰ型系统中cas3蛋白的解旋酶活性区域,使其成为只具备单链核酸酶活性的ncas3蛋白,达到在基因编辑过程中只在靶向位点切割其中一条dna单链而不会对dna分子内另一条单链造成损伤的目的。以此降低cas3蛋白在基因编辑过程中对细胞的毒性,从而提高转化效率,获得更多的转化子,进而筛选得到更多编辑后的目的菌株。
40.2、crispr
‑
cas系统的编辑效率与目标基因的分子量大小及其功能都有密切关系。一般来说,由于基因组损伤后修复的难易程度不同,更长基因在被编辑时,编辑质粒的转化效率相应更低,能够得到的转化子越少,其最终编辑的成功率也越低。目前,有关运动发酵单胞菌zm4内源i
‑
f型crispr
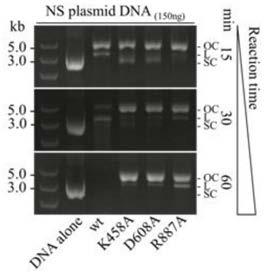
‑
cas系统的报道指出,该系统在敲除、替换单个基因时,可以达到近乎100%的效率。然而,在面对较大片段基因(约为基因组的5
‰
)时,其效率则仅能达到50%。
41.本技术中,通过比对zm4与其他同样带有内源i型crispr
‑
cas系统的菌株,内源crispr相关蛋白的编码基因序列差异,找到了其cas3蛋白的功能结构划分。并且通过探究i
‑
f型crispr
‑
cas系统的工作原理,设计并替换了zm4菌株内cas3蛋白解旋酶结构域中的一个氨基酸,使其丧失解旋酶活性,转变为只携带单链dna核酸内切酶活性的ncas3蛋白。
42.改造后的这一工程菌株命名为drm2菌株,并重新设计将其引用到敲除基因组大片段的实验当中。最终发现其在针对基因组大片段(约为基因组的5
‰
)编辑时,编辑质粒的转
化效率得到了显著提升(相比对照提升超过100倍),其最终的编辑效率也提高到接近100%。
附图说明
43.图1是实施例1中sds
‑
page电泳结果;
44.图2是实施例2中环状dna切割效果图;
45.图3是zm4菌株基因组上的cas3蛋白基因结构;
46.图4是实施例3中的菌落pcr产物cas3蛋白结构片段分析结果;
47.图5是实施例3中的菌落pcr产物电泳结果;
48.图6是实施例3中的pcr产物测序结果;
49.图7是实施例4中的工程菌株转化效率结果;
50.图8是实施例4中的工程菌株编辑效率结果。
具体实施方式
51.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例对本发明进行进一步详细说明,各实施例及试验例中所用的设备和试剂如无特殊说明,均可从商业途径得到。此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
52.根据本技术包含的信息,对于本领域技术人员来说可以轻而易举地对本发明的精确描述进行各种改变,而不会偏离所附权利要求的精神和范围。应该理解,本发明的范围不局限于所限定的过程、性质或组分,因为这些实施方案以及其他的描述仅仅是为了示意性说明本发明的特定方面。实际上,本领域或相关领域的技术人员明显能够对本发明实施方式作出的各种改变都涵盖在所附权利要求的范围内。
53.为了更好地理解本发明而不是限制本发明的范围,在本技术中所用的表示用量、百分比的所有数字、以及其他数值,在所有情况下都应理解为以词语“大约”所修饰。因此,除非特别说明,否则在说明书和所附权利要求书中所列出的数字参数都是近似值,其可能会根据试图获得的理想性质的不同而加以改变。各个数字参数至少应被看作是根据所报告的有效数字和通过常规的四舍五入方法而获得的。本发明中,“约”指给定值或范围的10%以内,优选为5%以内。
54.本发明下述各实施例中未特别限定温度时,则均为常温条件。常温是指四季中自然室温条件,不进行额外的冷却或加热处理,一般常温控制在10~30℃,最好是15~25℃。
55.本发明中涉及的基因、蛋白或其片段可以是天然纯化的产物,或是化学合成的产物,或使用重组技术从原核或真核宿主(例如,细菌、酵母、植物)中产生。
56.本发明披露了一种ncas3单链核酸内切酶及其应用,具体如下实施例所示。
57.实施例1蛋白质及其编码基因的制备与功能。
58.ncas3单链核酸内切酶是指在野生型cas3蛋白的解旋酶功能域上引入突变后,使其丧失解旋酶活性,从而转变为只具备单链核酸酶活性的ncas3蛋白,该单链核酸内切酶活性能以内切1,4
‑
磷酸二脂键的方式作用于靶标dna双链中的一条单链。
59.本发明本实施例中涉及的野生型(wide type)cas3蛋白的氨基酸序列如seq id no.1所示,核苷酸序列如seq id no.2所示。本实施例中具体共给出了三种不同序列的
ncas3单链核酸内切酶,分别命名为k458a(用丙氨酸取代野生型cas3蛋白第458位的赖氨酸,氨基酸序列与seq id no.1相比,区别仅在于将第458位的“k”替换为“a”;核苷酸序列与seq id no.2相比,区别仅在于将第1372
‑
1373位的“aa”替换为“gc”)、d608a(用丙氨酸取代野生型cas3蛋白第608位的天冬氨酸脯氨酸,氨基酸序列与seq id no.1相比,区别仅在于将第608位的“d”替换为“a”;核苷酸序列与seq id no.2相比,区别仅在于将第1823位的“a”替换为“c”)和r887a(用丙氨酸取代野生型cas3蛋白第887位的精氨酸,氨基酸序列与seq id no.1相比,区别仅在于将第887位的“r”替换为“a”;核苷酸序列与seq id no.2相比,区别仅在于将第2659
‑
2660位的“cg”替换为“gc”)。
60.1、模板dna的制备可以分别通过以下两种方式进行:
61.(1)运动发酵单胞菌zymomonas mobilis zm4总dna的提取:采用运动发酵单胞菌zymomonas mobilis zm4,取其新鲜湿菌体20克,悬于10毫升50mm tris缓冲液中(ph8.0),加入少量溶菌酶和8毫升0.25mm edta(ph8.0),混匀后37℃放置20min;之后加入2毫升质量分数为10%的sds,55℃放置5min,分别用等体积酚、氯仿各抽提一次;取最后一次的上清溶液,加入2倍体积乙醇,回收dna,分别用70%和无水乙醇洗;沉淀溶于0.5毫升te缓冲液(ph8.0,10mm tris,1mm edta),加入10mg/ml rnase 3μl,37℃保温1小时,分别用等体积酚、氯仿各抽提一次;上清溶液加入2倍体积乙醇,回收dna,分别用70%和无水乙醇洗,真空干燥,用去离子水溶解。
62.(2)人工合成三种ncas3单链核酸内切酶的核苷酸序列的dna片段。该合成过程在金斯瑞生物科技有限公司完成。
63.2、利用引物进行pcr扩增
64.根据目标核苷酸序列设计引物对如下:
65.正向引物:5
’‑
ctttaagaaggagatataccatatgaatgttctattcgtttcgc
‑3’
,如seq id no.3所示;
66.反向引物:5
’‑
gatctcagtggtggtggtggtggtgactatgatatctggaaaatc
‑3’
,如seq id no.4所示;
67.以运动发酵单胞菌zymomonas mobilis zm4的总dna或人工合成的ncas3单链核酸内切酶的核苷酸序列作为模板,用设计的引物对进行pcr扩增。
68.pcr反应体系:5μl 10
×
缓冲液;4μl dntp;0.5μl extaq dna聚合酶;1μl正向引物;1μl反向引物;0.5μl模板;38μl水。
69.pcr反应条件:94℃预变性5min,然后94℃变性30s,55℃退火1min30s,72℃延伸2min,30个循环,最后72℃延伸10min。
70.pcr产物用琼脂糖凝胶电泳检测产量和特异性,并用dna纯化试剂盒纯化。将纯化的pcr产物进行测序,结果表明pcr产物的序列符合预期,将突变序列命名为ncas3片段。
71.3、重组表达载体的构建
72.(1)将上述正确的pcr产物用琼脂糖凝胶电泳纯化回收。
73.(2)将质粒pet28a(cat.n0 69864
‑
3,novogen)用nco i和xho i双酶切,琼脂糖电泳回收酶切产物。
74.(3)将步骤(1)的回收产物和步骤(2)中的酶切产物进行连接,连接产物电击转化大肠杆菌dh5α后涂布于含有50μg/ml卡那霉素的lb平板,37℃过夜培养,将得到的转化子用
上述的正向引物和反向引物进行菌落pcr,筛选到含有ncas3基因的重组菌,提取重组菌的质粒,进行测序验证。结果表明,在pet28a的nco i和xho i酶切位点之间插入了ncas3基因片段,插入方向正确,将重组质粒命名为pet28a
‑
ncas3。
75.4、工程菌的制备
76.将质粒pet28a
‑
ncas3电击转化大肠杆菌bl21(de3)(cat.n0 cd801,全式金公司)后,涂布于含有50μg/ml卡那霉素的lb平板,37℃过夜培养,得到含有质粒pet28a
‑
ncas3的工程菌,记作bl21/pet28a
‑
ncas3。
77.用pet28a代替pet28a
‑
ncas3,转化大肠杆菌bl21(de3),步骤同上,得到含有pet28a的空载重组菌,作为对照菌。将转入pet28a的菌株记作bl21/pet28a。
78.5、ncas3单链核酸内切酶的制备与纯化
79.将上述步骤4中制备得到的阳性重组菌bl21/pet28a
‑
ncas3培养于含有50μg/ml卡那霉素的lb培养基中,37℃培养3h;od600=0.7时,加入iptg至其在lb培养基中的终浓度0.8mm,转至18℃继续培养16h。在3800rpm、15min条件下离心收集菌体,悬浮于pbs溶液(20mm tris
‑
hcl,ph7.4,0.5m nacl)中,于冰浴中超声破碎(60w,10min;超声1s,停止2s),之后12000rpm离心10min除去细胞碎片,取上清液;将上清液过his60 ni superflow resin纯化柱,用5ml超纯水冲洗,再用10ml溶液a(50mm nah2po4
‑
na2hpo4,ph7.0,25m m咪唑)漂洗,最后用5ml溶液b(50mm nah2po4
‑
na2hpo4,ph7.0,500mm咪唑)洗脱,收集洗脱液。然后将洗脱液用脱盐柱ge hitrap desalting进行脱盐处理,用溶液c(50mm tris
‑
hcl,ph 7.0)进行洗脱,得到的洗脱液再过阴离子交换柱ge hitrap q ff,先用溶液d(20mm tris
‑
hcl,ph7.9)洗脱杂蛋白,再用溶液e(20mm tris
‑
hcl,0.5m nacl,ph7.9)洗脱目的蛋白,得到ncas3单链核酸内切酶纯酶液。
80.将步骤4中制备的对照菌采用相同的步骤进行培养和纯化,得到的溶液作为对照酶液。
81.sds
‑
page电泳显示纯化的ncas3蛋白的分子量约为130kda,符合理论推断的130.0kda。结果如图1所示,图1中,泳道m表示蛋白分子量标准(180、130、95、72、55kda);泳道wt表示由野生型(wide type)cas3蛋白充当的阳性对照;泳道k458a、d608a、r887a表示在野生型cas3蛋白的解旋酶功能域上引入突变后,使其丧失解旋酶活性,从而转变为只具备单链核酸酶活性的ncas3蛋白;泳道ck表示由上述步骤4中的bl21/pet28a空载体表达纯化后得到的蛋白液,作为阴性对照。
82.实施例2以环状dna为底物验证蛋白功能
83.选取pl2r质粒(zheng et al,2019)作为反应底物,质粒总长度为3283bp。反应体系包含150ng pl2r环状质粒;2mm mgcl2;0.5mm atp;250nm cascade蛋白;250nm上述纯化得到的cas3或ncas3蛋白变体之一;一个携带靶向pl2r质粒上5
’‑
ccc
‑3’
pam序列的32nt的crrna。上述crrna由金斯瑞生物科技有限公司合成,序列如seq id no.5所示。在30℃条件下分别反应15、30、60分钟后,将反应产物跑琼脂糖凝胶电泳。
84.结果如图2所示,图2中最左侧泳道表示核酸分子量标准(5.0、3.0、2.0kb);最右侧reaction time轴表示反应持续时间(15、30、60min);右侧oc、l、sc表示反应过后环状dna分子的状态,[oc(open circle开环);l(linear线性);sc(negatively supercoiled超螺旋)];dna alone泳道表示反应体系中不加入cas3蛋白或ncas3蛋白时环状质粒的状态,随
着反应时间增加,质粒保持超螺旋状态不变,表示质粒dna结构未受到破坏;wt泳道表示反应体系中加入野生型(wide type)cas3蛋白时环状质粒的状态,随着反应时间增加,质粒逐渐由超螺旋转变为半开环再转变为线性最终直至消失,表示质粒dna在野生型cas3蛋白作用下先后发生了双链断裂,并逐渐被降解;k458a、d608a、r887a泳道表示反应体系中加入在野生型cas3蛋白的解旋酶功能域上引入突变后,使其丧失解旋酶活性,从而转变为只具备单链核酸酶活性的ncas3蛋白,随着反应时间增加,质粒逐渐由超螺旋状态转变为开环状态并且直至反应到最终时间仍保持开环状态并未向线性或降解发展,表示质粒dna在改造后的单链核酸酶ncas3蛋白作用下,只完成了dna分子中单链的断裂,结果符合实验预期。
[0085]
实施例3包含ncas3单链核酸内切酶的工程菌的制备
[0086]
本实施例中以d608a为例,制备包含ncas3单链核酸内切酶的工程菌。
[0087]
1、设计并构建单碱基编辑质粒
[0088]
(1)根据实验需求,决定利用zm4菌株中内源的i
‑
f型crispr
‑
cas编辑系统,对基因组上的目的位点进行编辑。
[0089]
如图3所示,通过对zm4菌株基因组上的cas3蛋白编码基因的仔细分析,发现cas3基因序列内存在5
’‑
tcc
‑3’
的pam序列,因此将其下游的32
‑
nt序列设计为一个原间隔序列。为了避免crispr
‑
cas系统破环转入的供体质粒,并且完成对zmo0681基因内的676,661号的腺嘌呤核苷酸到胞嘧啶核苷酸的原位替换,供体dna上总共引入了三种核苷酸变化。为了清晰说明,将原间隔序列的位置编号设定为:紧邻pam序列的下游位置称为1,随后位置称为2、3等,直至32;而pam序列中间的位置被称为
‑
1、
‑
2、
‑
3,其中
‑
1是最接近间隔序列的位置。于是三种核苷酸变化在供体dna上的位置即为c
‑
1t、c3t和t25g。其中c
‑
1t的和c3t分别破坏掉了原间隔序列中的pam序列和seed sequence序列,用于保护供体质粒,使编辑后的细胞得以存活。并且这两个位点的核苷酸替换并未造成对应氨基酸的变化,因此不会破坏原本的蛋白质序列。而t25g的突变则是为了将zmo0681基因内的676,661号的腺嘌呤核苷酸原位替换为胞嘧啶核苷酸,使位点对应的天冬氨酸(d)的密码子gat被替换为丙氨酸(a)的密码子gct。并且,为了使编辑后能够方便地筛选到阳性的转化子,c3t在破坏掉seed sequence的同时,引入了新的酶切位点dra i(tttaaa),使得后续可以通过对如图4所示的菌落pcr产物处理后的带型情况,快速筛选到具有预期编辑的菌株。
[0090]
同理,可针对k458a、r887a制备工程菌,分别为:
[0091]
将编码cas3蛋白的zmo0681基因内的676,209号的腺嘌呤核苷酸原位替换为鸟嘌呤核苷酸且将676,209号的腺嘌呤核苷酸原位替换为胞嘧啶核苷酸;
[0092]
将编码cas3蛋白的zmo0681基因内的677,497号的胞嘧啶核苷酸原位替换为鸟嘌呤核苷酸且将677,498号的鸟嘌呤核苷酸原位替换为胞嘧啶核苷酸。
[0093]
c
‑
1t和c3t两个碱基的改变属于密码子简并性的覆盖范围内,不影响蛋白质的翻译。只是为了原位替换时的操作,与工程菌性质无关。
[0094]
(2)按照(1)中的设计,如图3所示构建单碱基编辑质粒pns
‑
cas3。以质粒pl2r为基础,在上面分别引入原间隔序列s(d608)和含有三个指定核苷酸替换后的cas3。
[0095]
其中原间隔序列s由引物退火过程得到,将各1μl引物s(d608)
‑
f和引物s(d608)
‑
r混合到反应缓冲液中(10μl体系:1μl引物s(d608)
‑
f,1μl引物s(d608)
‑
r,1μl缓冲液,7μl水),经pcr仪95℃反应5分钟后再降至室温反应10分钟,得到原间隔序列s(d608),序列如
seq id no.16。
[0096]
s(d608)
‑
f:gaaagaatctttgcggggcgggcgacaaatcgcacc;seq id no.6;
[0097]
s(d608)
‑
r:gaacggtgcgatttgtcgcccgccccgcaaagattc;seq id no.7。
[0098]
核苷酸替换后的cas3基因序列(如seq id no.17所示)则是由zm4基因组在引物cas3
‑
f、d608a
‑
r、d608a
‑
f、cas3
‑
r(序列中带有下划线的部分分别为ecor i和xba i酶切位点)克隆得到。具体过程为:用cas3
‑
f和d608a
‑
r扩增出上半段up(d608a),用d608a
‑
f和cas3
‑
r扩增出下半段down(d608a),最后用cas3
‑
f和cas3
‑
r做overlap pcr将up(d608a)和down(d608a)连接为cas3(d608a)。
[0099]
cas3
‑
f:aggtcaccagctcaccgtctgaattcatgaatgttctattcgtttc;seq id no.8;
[0100]
d608a
‑
r:cttttaaatcataatcatccaattcagctaaaacgagatcagccccc;seq id no.9;
[0101]
d608a
‑
f:gctgaattggatgattatgatttaaaagatttacccgccttaactcg;seq id no.10;
[0102]
cas3
‑
r:ctcgagagatctgatatcactctagattaactatgatatctggaaa;seq id no.11。
[0103]
将质粒pl2r用bsa i核酸内切酶在37℃的条件下酶切4小时后,用琼脂糖凝胶电泳纯化回收。将回收产物与s(d608)通过t4 dna酶连技术组装连接。再将连接产物用ecor i和xba i核酸内切酶在37℃的条件下酶切4小时后,用琼脂糖凝胶电泳纯化回收。最后将回收产物与核苷酸替换后的cas3基因序列连接组装,得到单碱基编辑质粒pns
‑
cas3。
[0104]
2、利用编辑质粒构建工程菌株drm2
[0105]
(1)将上述构建完成的单碱基编辑质粒pns
‑
cas3通过热激转化的方式,转化到制备成感受态细胞的drm1菌株(zheng et al,2019)中。转化步骤如下:将质粒与感受态细胞冰上混合后,放入42℃水浴热激35s,然后加入1ml lb液体培养基,37℃培养1小时后离心收菌,涂带有壮观霉素抗性的平板。
[0106]
(2)将培养基上通过抗生素筛选过后的转化子收集,利用引物cas3
‑
chk
‑
f和cas3
‑
chk
‑
r进行菌落pcr。
[0107]
cas3
‑
chk
‑
f:gatcacggaaattatttggcttatggccttggtgctactgcgac;seq id no.12;
[0108]
cas3
‑
chk
‑
r:
[0109]
gaagacatccaaggcggcggcattaccgacaacatctatatcaaaattttc;seq id no.13。
[0110]
(3)在步骤(2)得到的pcr产物中,加入dra i限制性核酸内切酶(neb公司提供),酶切2小时后跑琼脂糖凝胶电泳。电泳结果如图5所示,其中最左侧泳道m表示核酸分子量标准(2.0、1.0、0.75kb);最右侧标记为胶图中核酸条带分子量;ck表示用drm1菌株基因组为模板,扩增后酶切得到的产物作为对照组;1、2、3、4泳道则为在平板上挑取得到的四个转化子。
[0111]
(4)将图5实验后得到的实际结果与图4预期得到的带型进行比较,发现泳道1、2、3对应的酶切后的带型都与预期编辑后应该产生的带型相符;而泳道4对应的带型则与对照组相同。表示泳道1、2、3对应的转化子内的基因组上完成了预期设计的核苷酸替换。
[0112]
(5)挑取其中的一株经过传代培养后,将其对应的pcr产物送到测序公司检测(由金斯瑞生物科技有限公司完成),测序结果如图6所示,峰图单一且与预期序列相符。drm2工程菌株构建成功。
[0113]
实施例4工程菌在大片段基因组敲除中的应用
[0114]
1、通过对zm4菌株基因组的分析,选取zm4基因组上的1,858,251
‑
1,868,309号基
因,即zmo1815
‑
zmo1822基因作为敲除目标,敲除片段序列共计10058个核苷酸。针对目标片段的基因组序列设计并构建了3个靶向目标区域内不同位点敲除质粒,即pko
‑
1、pko
‑
2和pko
‑
3(三个编辑质粒上都携带有一个人工crispr簇,和一段作为同源臂的donor序列。crispr簇中包含有针对于目标片段设计的间隔序列。三个质粒的区别只是在于间隔序列的不同,从而靶向目标区域内的不同位点。三个质粒的donor序列完全相同,都是在基因组上目标区域上下游各截取300bp序列组合而成)。其中每个敲除质粒上都携带有两个分别针对靶向序列dna编码链和模板链的间隔序列。
[0115]
2、将上述步骤中构建好的编辑质粒pko
‑
1、pko
‑
2和pko
‑
3通过电穿孔转化的方式,转入到drm2菌株中。电穿孔转化步骤如下:将质粒与drm2菌株感受态与冰上充分混合后,加入到直径为0.1cm的电转杯中。在电转仪中1.6kv条件下电转,加入rmg液体培养基复苏6小时后收菌涂带有rmg抗性平板。
[0116]
3、将上述步骤中电穿孔转化后平板放入30℃培养箱中培养48小时后,取出平板清点平板上转化子数量。由此计算可得每个编辑质粒转入drm2菌株时的转化效率,并与将它们转入drm1菌株时的转化效率相比较。
[0117]
转化效率结果如图7所示,图表表示将编辑质粒分别转化到drm1和drm2菌株的转化效率、与将空白载体(pez15a)转化到菌株中的转化效率的相对比值。其中wt表示将空白载体分别转化到drm1和drm2菌株中的转化效率,设为1.0。实验重复三次,误差线表示均值的标准误差。如图7结果所示,三个编辑质粒转化到drm2菌株中的转化效率相对于它们转化到drm1菌株中的转化效率都由显著提升,提升幅度在100倍左右。
[0118]
4、收集上述平板上的转化子,利用引物10k
‑
chk
‑
f和10k
‑
chk
‑
r进行菌落pcr。通过菌落pcr结果,可以判断目标片段是否成功被编辑质粒敲除。
[0119]
10k
‑
chk
‑
f:gacaagagcggaatccgcgt;seq id no.14
[0120]
10k
‑
chk
‑
r:gaggtaataaccccgcgacc;seq id no.15。
[0121]
统计编辑效率,结果如图8所示,三个编辑质粒转化到drm2菌株后,其针对目标位点的基因编辑效率,相对于它们转化到drm1菌株后的编辑效率均有显著提升,甚至最高达到了100%。证明drm2菌株在用于针对大片段基因组敲除时,可以有效提高编辑质粒得转化效率,得到更多转化子得同时,进而大幅提高编辑效率。
[0122]
k458a、r887a和d608a虽然是三个不同的突变位点,但是达成的目的都是沉默掉了野生型菌株中cas3蛋白的解旋酶功能域,从而使其转变为具备单链切割活性的核酸内切酶。技术效果(提高编辑质粒的转化效率、成功对大片段基因组进行敲除)的根本原理,正是利用改造后cas3蛋白的单链切割特点,区别于传统的直接将dna双链切断,而是只留下一个切口。减少对待编辑菌株基因组的损伤,从而提高转换效率,使得对本来较难实现的大片段的敲除成为可能。因此,在d608a采用基因组原位替换构建的工程菌可以提高编辑质粒的转化效率、成功对大片段基因组进行敲除,可以推断采用相同原理针对k458a和r887a构建的工程菌株比如也可以达到相同的技术效果。
[0123]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

