一种依度沙班手性叠氮中间体化合物的制备方法
(一)技术领域
1.本发明涉及一种依度沙班手性叠氮中间体化合物的制备方法。
(二)
背景技术:
2.依度沙班是日本第一三共株式会社颜值的小分子口服抗凝药。2011年4月获得批准,2011年7月在日本上市。凝血过程中,活化的凝血因子x(fxa)将凝血酶原(fii)激活成为凝血酶(fiia),促使纤维蛋白形成,由此形成血栓,因而fxa已成为新一代抗凝血药物的主要八点。依度沙班通过选择性、可逆性的抑制fxa达到抑制血栓形成的目的。临床研究验证了其良好的安全性、口服吸收性以及抗凝血活性。依度沙班的化学结构如图1所示。
3.目前,依度沙班的制备主要是有三个片段a、b、c通过构建酰胺键合成,其中片段a是合成路线中的关键中间体,带有3个手性中心,制备路线如图2所示。
4.在该路线中,从中间体04到中间体06的转化,现有文献(wo2014081047、ep1925611、ep2407450、ep2407457、ep2589590、us20150353577、jp5801011以及org.process res.dev.2019,23(4),524
‑
534等)中均使用了nan3这一高度易爆的危险品,限制了化合物的工业制备。
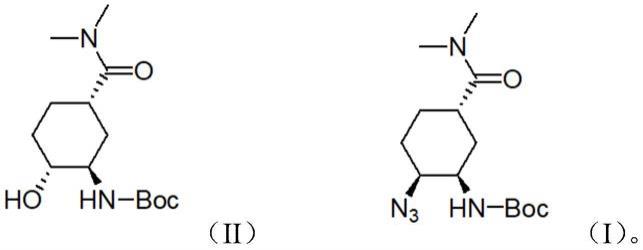
5.以上现有技术中还存在以下缺点:(1)在中间体04(即化合物(ii))到中间体05的制备中,使用了甲磺酰氯对羟基进行保护,而甲磺酰氯作为剧毒品,在购买、储存、使用等多方面都存在着不便;(2)在中间体05到中间体06(即化合物(i))的制备中,采用nan3对甲磺酰基进行取代,专利us2012053349a1描述该反应消去副产物多、产物手性选择性较差且产生的手性异构体很难分离,目标化合物的手性纯度低;(3)叠氮化钠大大过量,含有nan3的有机溶剂和废水不易处理,不利于环境友好。
(三)
技术实现要素:
6.本发明目的是提供一种安全风险低、适合工业化生产、高效率的依度沙班手性叠氮中间体化合物的制备方法。
7.本发明采用的技术方案是:
8.一种依度沙班手性叠氮中间体化合物的制备方法,所述方法是以式(ii)所示羟基化合物为底物,在脒类结构化合物存在下,与有机叠氮类化合物在有机溶剂中进行取代反应,制得式(i)所示手性叠氮中间体化合物;
9.10.具体的,所述有机叠氮类化合物为下列之一:叠氮磷酸二苯酯、对甲苯磺酰叠氮、叠氮基三甲基硅烷。
11.本发明有机叠氮化合物作为叠氮基团的来源,避免使用nan3这一高度易爆的危险品,更为安全。而且,本发明方法收率可以达到85%以上,远高于现有技术的23.9~68.1%%(来自于专利ep2407450、wo2007032498等)。该方法也可以对手性进行良好的控制,并得到构型完全反转的目标化合物,该化合物经过钯炭加氢还原后所得粗品进行重结晶,得到的二胺化合物手性纯度可以达到几乎100%。
12.优选的,所述脒类结构化合物为下列之一:甲咪、dbn、dbu、三氮脒。
13.优选的,所述有机溶剂为下列之一或其中两种以上的混合物:四氢呋喃、2
‑
甲基四氢呋喃、甲基叔丁基醚、甲苯、二氯甲烷、甲基异丁基酮。
14.优选的,所述羟基化合物、有机叠氮化合物、脒类结构化合物物质的量用量之比为1:1~2:1~2。
15.优选的,所述取代反应在
‑
20℃~50℃下进行。
16.优选的,所述反应在氮气保护条件下进行。
17.具体的,所述方法如下:在氮气保护下,往反应容器中依次加入式(ii)所示羟基化合物、有机叠氮类化合物和有机溶剂,冷却到0
±
5℃,加入脒类结构化合物,在0
±
5℃反应1~2小时,然后恢复到室温继续反应12~18小时,反应结束后,反应体系依次用水、3~5%盐酸溶液洗涤,然后减压浓缩,柱纯化得到式(i)所示手性叠氮中间体化合物。
18.本发明的有益效果主要体现在:本发明将两步合成反应直接优化成一步反应合成,采用叠氮磷酸二苯酯、对甲苯磺酰叠氮、叠氮基三甲基硅烷等有机叠氮化合物作为叠氮基团的来源,在甲咪、dbn、dbu、三氮脒等脒类结构化合物的协助下,在适当的溶剂中发生取代反应,得到目标化合物手性叠氮中间体;不仅成功的避免了剧毒品甲磺酰氯、易爆品叠氮化钠的使用,且降低了工艺难度和生产成本。
(四)附图说明
19.图1为依度沙班的化学结构图;
20.图2为依度沙班的制备路线图;
21.图3为实施例1制得式(i)所示手性叠氮中间体化合物的纯度hplc图;
22.图4为实施例1制得式(i)所示手性叠氮中间体化合物的手性纯度分析hplc图。
(五)具体实施方式
23.下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
24.实施例1:
25.50ml反应瓶中,氮气保护条件下,依次加入式(ii)所示羟基化合物(3.0g,10.5mmol)、叠氮磷酸二苯酯(cas:26386
‑
88
‑
9)(3.5g,12.7mmol),甲苯(18ml)。冷却到0℃。加入甲脒(0.56g,12.7mmol)。反应在0℃反应2小时,然后恢复到室温继续反应16小时。液相跟踪中间体04含量低于0.2%。体系依次用水10ml*2、5%(w/w)盐酸10ml洗涤后减压浓缩、柱纯化agela flash column silica
‑
cs(40g)得到2.89g(收率88.5%)式(i)所示手性叠氮
中间体化合物(纯度98.5%),其纯度hplc图参见图3,手性纯度分析hplc图参见图4。
26.核磁数据:1h
‑
nmr(cdcl3)δ:1.45(9h,s),1.58
‑
1.66(1h,m),1.67
‑
1.76(1h,m),1.84
‑
1.96(2h,m),2.04
‑
2.15(1h,m),2.17
‑
2.26(1h,m),2.75
‑
2.81(1h,m),2.94(3h,s),3.04(3h,s),3.07(3h,s),4.00
‑
4.08(1h,m),4.69
‑
4.82(2h,m).
27.钯炭加氢还原:取1g式(i)所示手性叠氮中间体化合物(纯度98.5%),加入10ml甲醇,搅拌溶清,加入10%钯碳0.05g(含水量50%),加热至40℃,通氢气1小时,降温,抽滤,浓缩,所得浓缩物用两倍乙腈重结晶,得到二胺化合物0.8g(手性纯度99.99%)。
[0028][0029]
实施例2:
[0030]
50ml反应瓶中,氮气保护条件下,依次加入式(ii)所示羟基化合物(3.0g,10.5mmol)、叠氮磷酸二苯酯(3.5g,12.7mmol),甲苯(18ml)。冷却到0℃。加入dbu(1.93g,12.7mmol)。反应在0℃反应2小时,然后恢复到室温继续反应16小时。液相跟踪中间体04含量低于0.2%。体系依次用水10ml*2、5%盐酸10ml洗涤后减压浓缩、柱纯化得到2.97g(收率91%)式(i)所示手性叠氮中间体化合物(纯度98.9%)。
[0031]
实施例3:
[0032]
50ml反应瓶中,氮气保护条件下,依次加入中间体04式(ii)所示羟基化合物(3.0g,10.5mmol)、叠氮磷酸二苯酯(3.5g,12.7mmol),无水四氢呋喃(18ml)。冷却到0℃。加入dbu(1.93g,12.7mmol。反应在0℃反应2小时,然后恢复到室温继续反应16小时。液相跟踪中间体04含量低于0.2%。加入20ml水和20ml甲苯,搅拌15分钟后静置分层。有机相体系依次用水10ml*2、5%盐酸10ml洗涤后减压浓缩柱纯化得到2.90g(收率89%)式(i)所示手性叠氮中间体化合物(纯度98.7%)。
[0033]
实施例4:
[0034]
50ml反应瓶中,氮气保护条件下,依次加入式(ii)所示羟基化合物(3.0g,10.5mmol)、叠氮磷酸二苯酯(3.5g,12.7mmol),甲基叔丁基醚(18ml)。冷却到0℃。加入dbu(1.93g,12.7mmol。反应在0℃反应2小时,然后恢复到室温继续反应16小时。液相跟踪中间体04含量低于0.2%。体系依次用水10ml*2、5%盐酸10ml洗涤后减压浓缩柱纯化得到2.90g(收率89%)式(i)所示手性叠氮中间体化合物(纯度98.4%)。
[0035]
实施例5:
[0036]
50ml反应瓶中,氮气保护条件下,依次加入式(ii)所示羟基化合物(3.0g,10.5mmol)、叠氮磷酸二苯酯(3.5g,12.7mmol),甲基叔丁基醚(18ml)。冷却到0℃。加入dbn(1.58g,12.7mmol。反应在0℃反应2小时,然后恢复到室温继续反应16小时。液相跟踪中间
体04含量低于0.2%。体系依次用水10ml*2、5%盐酸10ml洗涤后减压浓缩柱纯化得到2.80g(收率86%)式(i)所示手性叠氮中间体化合物(纯度98.2%)。
[0037]
实施例6:
[0038]
250ml反应瓶中,氮气保护条件下,依次加入式(ii)所示羟基化合物(30g,0.105mol)、叠氮磷酸二苯酯(35g,0.127mmol),甲苯(180ml)。冷却到0℃。加入dbu(19.3g,0.127mol。反应在0℃反应2小时,然后恢复到室温继续反应16小时。液相跟踪中间体04含量低于0.2%。体系依次用水100ml*2、5%盐酸100ml洗涤后减压浓缩柱纯化得到29.4g(收率90%)式(i)所示手性叠氮中间体化合物(纯度98.8%)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

