一种1,1,1,3,3
‑
五氟丙烷的制备方法
技术领域
1.本技术涉及一种1,1,1,3,3
‑
五氟丙烷的制备方法,属于化学品制备技术领域。
背景技术:
2.1,1,1,3,3
‑
五氟丙烷(hfc
‑
245fa)被广泛用作发泡剂、制冷剂、清洗剂、热传导介质、气溶胶推进剂等。hfc
‑
245fa的odp值为0、gwp值较低、不燃、无毒,且具有与一氟二氯乙烷(hchc
‑
141b)和一氟三氯甲烷(cfc
‑
11)相似的性质,对普通abs板无腐蚀作用,不需改变发泡生产设备即可使用,作为hchc
‑
141b和cfc
‑
11的良好替代品,在发泡剂工业中受到广泛的应用。
3.hfc
‑
245fa的各种合成工艺中由1,1,1,3,3
‑
五氯丙烷(hcc
‑
240fa)氟化合成hfc
‑
245fa的工艺具有原料易得,反应步骤少,工艺简单等优点,较适合于工业化生产。以hcc
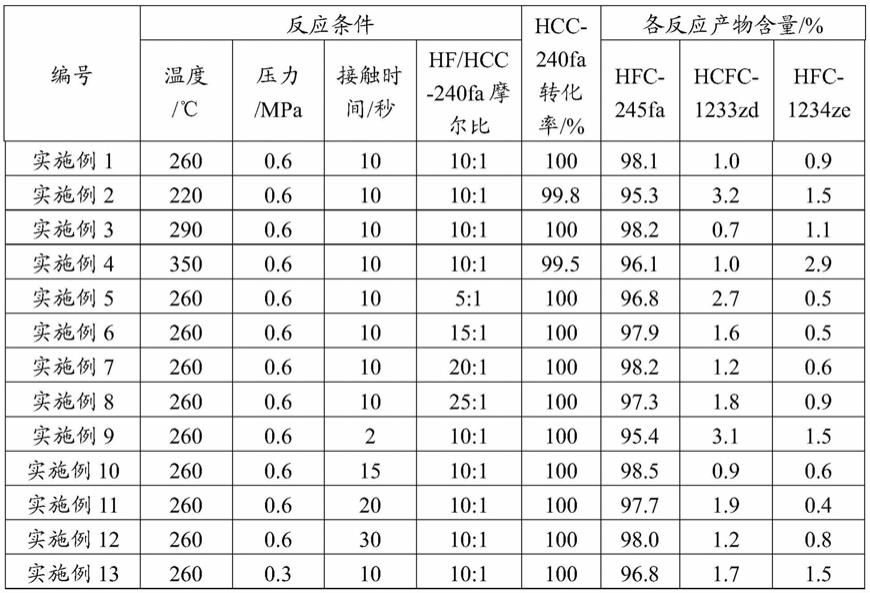
‑
240fa为原料制备hfc
‑
245fa的工艺分为液相氟化工艺、气相氟化工艺。液相氟化工艺通过一步氟化即可生成hfc
‑
245fa,气相氟化工艺合成hfc
‑
245fa需经过二或三个反应步骤,首先hcc
‑
240fa和hf反应生成1
‑
氯
‑
3,3,3
‑
三氟丙烯(hcfc
‑
1233zd)、1,3,3,3
‑
四氟丙烯(hfc
‑
1234ze)和少量的hfc
‑
245fa的混合物,然后hcfc
‑
1233zd和hfc
‑
1234ze在随后的步骤进一步氟化成预期产物hfc
‑
245fa。
4.中国专利cn1130614a公开了通过液相法采用sbcl5催化剂制备hfc
‑
245fa的工艺,该工艺虽然具有反应温度低、能耗小等优点,但其对环境污染和设备腐蚀严重。
5.中国专利cn101028993a公开了一种1,1,1,3,3
‑
五氟丙烷的制备方法,以氟化氢和hcc
‑
240fa为原料,经hcfc
‑
1233zd、1,1,1,3
‑
四氟丙烯(hfc
‑
1234ze)中间体三步气相催化氟化反应得到1,1,1,3,3
‑
五氟丙烷。第一反应器主要进行氟化hcc
‑
240fa合成hcfc
‑
1233zd,第二反应器主要进行氟化hcfc
‑
1233zd合成hfc
‑
1234ze,第三反应器主要进行氟化hfc
‑
1234ze合成hfc
‑
245fa。该方法的不足之处是制备路线较长,设备投资大,反应能耗高。
6.中国专利cn100546959c公开了一种1,1,1,3,3
‑
五氟丙烷的生产方法,在铬基催化剂存在下,将1,1,1,3,3
‑
五氯丙烷和无水氟化氢,通过两步气相催化氟化反应生产hfc
‑
245fa,第一步反应生成物hcfc
‑
1233zd、hfc
‑
1234ze、少量目的产物hfc
‑
245fa、副产物hcl和未反应的原料经“hcl分离塔”除去hcl后,再经原料分离塔分出hcc
‑
240fa返回第一步反应,其余有机物料进入第二步反应。该路线对两步反应的产物分别进行分离,步骤复杂,能量效率低、设备投资大。
技术实现要素:
7.本发明的目的是提供一种以1,1,1,3,3
‑
五氯丙烷和无水氟化氢为原料一步气相法制备及分离获得1,1,1,3,3
‑
五氟丙烷的方法,该方法反应步骤少,设备投资小,节能环保,且1,1,1,3,3
‑
五氟丙烷的收率达到了99.5wt%以上。
8.为了实现上述目的,本发明提供了一种1,1,1,3,3
‑
五氟丙烷的制备方法,包括以下步骤:
9.(a)1,1,1,3,3
‑
五氯丙烷和氟化氢在氟化催化剂的作用下气相氟化,得到反应生成的1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯、1,3,3,3
‑
四氟丙烯、氯化氢和未反应的氟化氢的混合物;
10.(b)将步骤(a)中得到的混合物引入分离塔,塔顶分离出氯化氢,塔釜得到1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯、1,3,3,3
‑
四氟丙烯和氟化氢混合物,除酸后进入粗品罐;
11.(c)将所述粗品罐中的混合物引入脱气塔,塔顶分离出1,3,3,3
‑
四氟丙烯返回至反应器与氟化氢反应生成1,1,1,3,3
‑
五氟丙烷,塔釜得到1,1,1,3,3
‑
五氟丙烷和1
‑
氯
‑
3,3,3
‑
三氟丙烯混合物;
12.(d)将步骤(c)中塔釜混合物引入精馏塔,得到1,1,1,3,3
‑
五氟丙烷。
13.优选地,所述步骤(a)中反应温度为220℃~350℃,更优选250℃~330℃;反应压力为0.3mpa~1.3mpa,更优选0.5mpa~1.2mpa;反应物的接触时间为2~30秒,更优选5~20秒。
14.优选地,所述步骤(a)中氟化氢与1,1,1,3,3
‑
五氯丙烷的摩尔比为(5~25):1,更优选(10~20):1。
15.优选地,所述步骤(a)中氟化催化剂为氧化铬、氟化铬、氟化氧化铬、氟化铝、氟化氧化铝、负载氧化铬的氟化铝、活性炭、氟化镁和负载金属离子的铬基氟化催化剂中的至少一种。
16.优选地,所述步骤(a)中氟化催化剂为负载金属离子的铬基氟化催化剂,所述金属的氧化价态大于或等于3。
17.优选地,所述氧化价态大于或等于3的金属选自铑、锑、钽、铌、镁、钛、锆、钼、钒或锡中的一种或多种,更优选锑、钛、镁和锡中的一种或多种。
18.优选地,将步骤(c)中塔釜混合物引入萃取精馏塔,塔顶分离出1
‑
氯
‑
3,3,3
‑
三氟丙烯返回至反应器脱除氯化氢生成1,3,3,3
‑
四氟丙烯,所述1,3,3,3
‑
四氟丙烯与所述氟化氢反应生成所述1,1,1,3,3
‑
五氟丙烷,塔釜得到1,1,1,3,3
‑
五氟丙烷和萃取剂的混合物;
19.将所述塔釜得到1,1,1,3,3
‑
五氟丙烷和萃取剂的混合物引入萃取剂回收塔,塔顶分离出所述1,1,1,3,3
‑
五氟丙烷,塔釜得到的萃取剂返回至所述萃取精馏塔。
20.优选地,所述萃取剂为环戊烷、正戊烷、四氯化碳和丙酮中的一种或多种,更优选环戊烷与丙酮的混合物。
21.优选地,将所述步骤(c)中塔釜混合物引入共沸精馏塔,塔顶分离出1
‑
氯
‑
3,3,3
‑
三氟丙烯和1,1,1,3,3
‑
五氟丙烷的共沸物返回至反应器,所述1
‑
氯
‑
3,3,3
‑
三氟丙烯脱除氯化氢生成1,3,3,3
‑
四氟丙烯,所述1,3,3,3
‑
四氟丙烯与所述氟化氢反应生成所述1,1,1,3,3
‑
五氟丙烷,塔釜富集的1,1,1,3,3
‑
五氟丙烷进入产品后处理系统,经除酸、脱水得到所述1,1,1,3,3
‑
五氟丙烷。
22.优选地,所述1,1,1,3,3
‑
五氟丙烷的收率为99.5wt%以上。
23.优选地,所述反应器为列管式固定床反应器。
24.本技术的有益效果包括但不限于:
25.本技术的1,1,1,3,3
‑
五氟丙烷是以1,1,1,3,3
‑
五氯丙烷和无水氟化氢为原料通过一步气相法制备及分离后得到,1,1,1,3,3
‑
五氯丙烷的转化率达到了100%,1,1,1,3,3
‑
五氟丙烷的收率达到了99.5wt%以上;1,1,1,3,3
‑
五氟丙烷的制备方法通过优化反应和分离过程的工艺参数,使得反应步骤减少,设备投资降低,节能环保,同时降低了生产成本;不同的精馏方法适用于不同的生产环境,有利于大规模的工业化生产。
附图说明
26.此处所说明的附图用来提供对本技术的进一步理解,构成本技术的一部分,本技术的示意性实施例及其说明用于解释本技术,并不构成对本技术的不当限定。在附图中:
27.图1为本技术中1,1,1,3,3
‑
五氟丙烷的制备流程示意图。
具体实施方式
28.下面结合实施例详述本技术,但本技术并不局限于这些实施例。
29.在一个示例中,如图1所示,图中附图标记1、2、4、6、7、9、11、12、14、15、17和18均为管线,3为反应器,5为hcl分离塔,8为粗品罐,10为脱气塔,13为萃取精馏塔,16为萃取剂回收塔。
30.hf和hcc
‑
240fa预热后经管线1、2进入反应器3,在氟化催化剂的作用下进行反应,得到的产物流经管线4进入hcl分离塔5,塔顶组分氯化氢通过管线6进入制酸系统制得盐酸。塔釜组分1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯、1,3,3,3
‑
四氟丙烯和氟化氢经过水碱洗脱除酸度,经过管线7进入粗品罐8。粗品罐8中的物料经管线9进入脱气塔10,塔顶组分1,3,3,3
‑
四氟丙烯通过管线11返回反应器3进行反应,塔釜组分1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯通过管线12进入进入萃取精馏塔13,塔顶组分1
‑
氯
‑
3,3,3
‑
三氟丙烯通过管线14返回反应器3继续反应,塔釜组分1,1,1,3,3
‑
五氟丙烷和萃取剂经过管线15进入萃取剂回收塔16,塔顶经过管线17得到目标产物1,1,1,3,3
‑
五氟丙烷,塔釜萃取剂通过管线18循环至萃取精馏塔13。
31.本示例中反应分析方法如下:
32.在内径为38mm的碳钢管中加入50毫升含有锑、钛、镁和锡的铬基氟化催化剂,将反应器升温至220℃~350℃,通入预热的hf和hcc
‑
240fa进行反应,控制hf与hcc
‑
240fa的摩尔比为(5~25):1,接触时间为2~30秒,反应压力0.3mpa~1.3mpa,反应20h后,反应产物经水洗、碱洗、碱干燥除去hcl和hf后,用气相色谱分析反应产物的组成,结果如表1所示。
33.表1一步气相反应的条件及各产物组成
[0034][0035][0036]
从表1可以看出,反应条件优化之后,反应步骤减少,降低了生产成本,反应更加节能、环保、高效,实施例1~15中hfc
‑
245fa在未进行分离之前收率就在95wt%以上;与实施例1相比,对比例1中hcc
‑
240fa的转化率和hfc
‑
245fa的含量较低,说明气相反应温度不宜过高;对比例2、3和4中hfc
‑
245fa的含量较低,说明气相反应压力应设置在合理的范围内,接触时间不宜过长,hf和hcc
‑
240fa摩尔比也应控制在合理范围内。
[0037]
本示例中分离方法如下:
[0038]
首先,将反应生成的1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯、1,3,3,3
‑
四氟丙烯、氯化氢和未反应的氟化氢的混合物引入分离塔,塔顶分离出氯化氢,塔釜得到1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯、1,3,3,3
‑
四氟丙烯和氟化氢混合物,除酸后进入粗品罐;
[0039]
随后,将粗品罐中的混合物引入脱气塔,塔顶分离出1,3,3,3
‑
四氟丙烯返回至反应器与氟化氢反应生成1,1,1,3,3
‑
五氟丙烷,塔釜得到1,1,1,3,3
‑
五氟丙烷和1
‑
氯
‑
3,3,3
‑
三氟丙烯混合物;
[0040]
然后,将塔釜得到1,1,1,3,3
‑
五氟丙烷和1
‑
氯
‑
3,3,3
‑
三氟丙烯混合物引入萃取精馏塔,塔顶分离出1
‑
氯
‑
3,3,3
‑
三氟丙烯返回至反应器脱除氯化氢生成1,3,3,3
‑
四氟丙烯,1,3,3,3
‑
四氟丙烯与氟化氢反应生成1,1,1,3,3
‑
五氟丙烷,塔釜得到1,1,1,3,3
‑
五氟丙烷和萃取剂的混合物;
[0041]
最后,将塔釜得到1,1,1,3,3
‑
五氟丙烷和萃取剂的混合物引入萃取剂回收塔,塔顶分离出1,1,1,3,3
‑
五氟丙烷,塔釜得到的萃取剂返回至萃取精馏塔循环利用;
[0042]
其中,萃取剂为环戊烷与丙酮的混合物。
[0043]
在另一个示例中,分离方法如下:
[0044]
首先,将反应生成的1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯、1,3,3,3
‑
四氟丙烯、氯化氢和未反应的氟化氢的混合物引入分离塔,塔顶分离出氯化氢,塔釜得到1,1,1,3,3
‑
五氟丙烷、1
‑
氯
‑
3,3,3
‑
三氟丙烯、1,3,3,3
‑
四氟丙烯和氟化氢混合物,除酸后进入粗品罐;
[0045]
随后,将粗品罐中的混合物引入脱气塔,塔顶分离出1,3,3,3
‑
四氟丙烯返回至反应器与氟化氢反应生成1,1,1,3,3
‑
五氟丙烷,塔釜得到1,1,1,3,3
‑
五氟丙烷和1
‑
氯
‑
3,3,3
‑
三氟丙烯混合物;
[0046]
将塔釜得到1,1,1,3,3
‑
五氟丙烷和1
‑
氯
‑
3,3,3
‑
三氟丙烯混合物引入共沸精馏塔,塔顶分离出1
‑
氯
‑
3,3,3
‑
三氟丙烯和1,1,1,3,3
‑
五氟丙烷的共沸物返回至反应器,1
‑
氯
‑
3,3,3
‑
三氟丙烯脱除氯化氢生成1,3,3,3
‑
四氟丙烯,1,3,3,3
‑
四氟丙烯与氟化氢反应生成1,1,1,3,3
‑
五氟丙烷,塔釜富集的1,1,1,3,3
‑
五氟丙烷进入产品后处理系统,经除酸、脱水得到1,1,1,3,3
‑
五氟丙烷。
[0047]
两个示例中,1,1,1,3,3
‑
五氟丙烷的收率均为99.5wt%以上。
[0048]
以上所述,仅为本技术的实施例而已,本技术的保护范围并不受这些具体实施例的限制,而是由本技术的权利要求书来确定。对于本领域技术人员来说,本技术可以有各种更改和变化。凡在本技术的技术思想和原理之内所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

