基于d
‑
a共轭聚合物半导体的室温类stille交叉偶联共聚反应
技术领域:
1.本发明涉有机半导体活性层给体
‑
受体(d
‑
a)共轭聚合物合成的技术领域,具体涉及一种基于价廉的芳基二硫醚和芳基二锡烷的交叉偶联室温聚合方法。
背景技术:
2.有机共轭聚合物由于其在塑料电子中的巨大潜力而引起了人们的注意,如化学结构可调节性、带隙和光吸收可调控、电荷传输迁移率优良、可柔性制备和溶液加工性能等。特别是无缺陷、结构完美交替的π
‑
共轭d
‑
a共聚物,比存在结构缺陷的d
‑
a共聚物,电荷传输性能更加优异。
3.d
‑
a共轭聚合物传统的制备方法,是以传统的芳基二卤化物作为亲电试剂,以四(三苯基膦)钯(pd(pph3)4)为催化剂,与芳基二锡烷在加热和氮气保护条件下进行stille交叉偶联聚合。但芳基卤化物在自然界中存在较少,通常需要多一步卤化的反应过程,这样难以实现原子经济的特点。因此,人们也在探索能用自然界大量存在的物质来实现交叉偶联聚合的方法。
4.刚刚上面提到的stille交叉偶联是在加热条件下进行的,可是在该条件下不仅增大了反应所需的能耗,更有可能会使共聚物产生结构缺陷。而结构缺陷的产生,会使d
‑
a共聚物的电荷迁移率降低。因而如何在室温条件下实现无缺陷或者低缺陷的共聚物,成了有机半导体领域里亟待解决的问题。
技术实现要素:
5.针对上述暴露的问题,本发明提供了一种以芳基二硫醚为亲电试剂,以芳基二锡烷为亲核试剂的类stille交叉偶联聚合方法,该方法以噻吩
‑2‑
甲酸亚铜(cutc)和pd(pph3)4为催化剂,在常温条件下实现类stille交叉偶联共聚反应。本发明是基于缺电子芳基二硫醚为亲电试剂与芳基二锡烷为亲核试剂的类stille交叉偶联共聚反应。反应通式如下:
[0006][0007]
其中,芳基二硫醚为下列任意一种化合物:
[0008]
[0009][0010]
r,r1,r2,r3为独立的氢或c1‑
c
30
的烷基,其中包括碳原子总数为6
‑
16的直链烷基包括:正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基;以及碳原子总数为8
–
30的支链烷基包括:2
‑
乙基己基,2
‑
乙基辛基,2
‑
丁基己基、2
‑
己基辛基、4
‑
己基癸基、3
‑
己基十一烷基、2
‑
辛基癸基、2
‑
辛基十二烷基、3
‑
辛基十三烷基、2
‑
癸基十二烷基、2
‑
癸基十四烷基、3
‑
癸基十五烷基、2
‑
十二烷基十六烷基、4
‑
辛基十四烷基、4
‑
癸基十六烷基、4
‑
己基癸基、4
‑
辛基十二烷基、4
‑
癸基十四烷基、4
‑
十二烷基十六烷基。
[0011]
芳基二锡烷为下列任意一种化合物:
[0012]
[0013][0014]
r’,r1’
,r2’
,r3’
为独立的氢或c1‑
c
30
的烷基,其中包括碳原子总数为6
‑
16的直链烷基包括:正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基;以及碳原子总数为8
–
30的支链烷基包括:2
‑
乙基己基,2
‑
乙基辛基,2
‑
丁基己基、2
‑
己基辛基、4
‑
己基癸基、3
‑
己基十一烷基、2
‑
辛基癸基、2
‑
辛基十二烷基、3
‑
辛基十三烷基、2
‑
癸基十二烷基、2
‑
癸基十四烷基、3
‑
癸基十五烷基、2
‑
十二烷基十六烷基、4
‑
辛基十四烷基、4
‑
癸基十六烷基、4
‑
己基癸基、4
‑
辛基十二烷基、4
‑
癸基十四烷基、4
‑
十二烷基十六烷基。
[0015]
在本发明的方法中,其中所述的催化剂包括有机钯催化剂,优选为pd(pph3)4,pd/c,pdcl2,pd(oac)2的任意一种或其组合。
[0016]
在本发明的方法中,其中所述的催化剂还包括cutc。
[0017]
在本发明的方法中,其中所述的催化剂为pd(pph3)4/cutc。
[0018]
在本发明的方法中,其中所述的溶剂选自乙醇、氯仿,四氯化碳,甲苯,丙酮,thf,dmso中的任意一种或其组合。
[0019]
在本发明的方法中,其特征在于,所述方法步骤包括:
[0020]
(1)采用惰性气体保护,在pd(pph3)4和cutc共同的催化作用下,将芳基二硫醚和芳基二锡烷置于甲苯中在温室条件下进行反应,得到混合物;
[0021]
(2)将得到的混合物滴入甲醇中,析出固体,采用索氏提取器对析出固体进行索氏提取,将经索氏提取得到的聚合物溶液用氨水洗过之后再进行浓缩,然后将经浓缩得到的溶液滴入甲醇中,析出的固体为具有超过10个重复单元的高分子量的d
‑
a共轭聚合物。
[0022]
本发明是基于芳基二硫醚与芳基二锡烷在pd(pph3)4和cutc共同催化作用下实现的,具体合成方法为:
[0023]
将1当量的芳基二硫醚、1当量的芳基二锡烷、5当量cutc和pd(pph3)4(10mol)%(缺电子的芳基二锡烷还需要加入zn(oac)2(10mol%))放入到反应瓶中。置换氮气三次后,加入干燥的甲苯(使溶液的浓度保持在0.025m)。室温搅拌24小时后(缺电子的芳基二锡烷需要搅拌72小时),加入甲醇使沉淀析出。过滤之后,将滤饼依次分别用丙酮、正己烷、氯仿、氯苯进行索氏提取。氯仿和氯苯的提取液用浓氨水洗过之后,将聚合物溶液浓缩,然后用甲醇析出沉淀。沉淀过滤、干燥之后,即为目标聚合物。
[0024]
本发明的优点在于:
[0025]
(1)以芳基硫醚为底物作为亲电试剂,相对于传统的芳基卤代物作为亲电试剂,芳
基硫醚在自然界中大量存在。具有原子经济的优点。
[0026]
(2)反应在室温条件下进行,反应所需的能耗低,可以大大地降低成本。
[0027]
(3)聚合反应所产生的缺陷,相对于芳基卤代物作为亲电试剂的聚合反应要小很多。
附图说明:
[0028]
图1为上述8个实施举例聚合物在氯仿溶液中的紫外
‑
可见吸收光谱。
[0029]
图2为上述8个实施举例聚合物在薄膜状态下的紫外
‑
可见吸收光谱。
[0030]
图3为上述8个实施举例聚合物的热重分析曲线。
[0031]
图4为聚合物p4顶栅底接触otfts器件的转移曲线。
[0032]
图5为聚合物p5顶栅底接触otfts器件的转移曲线。
[0033]
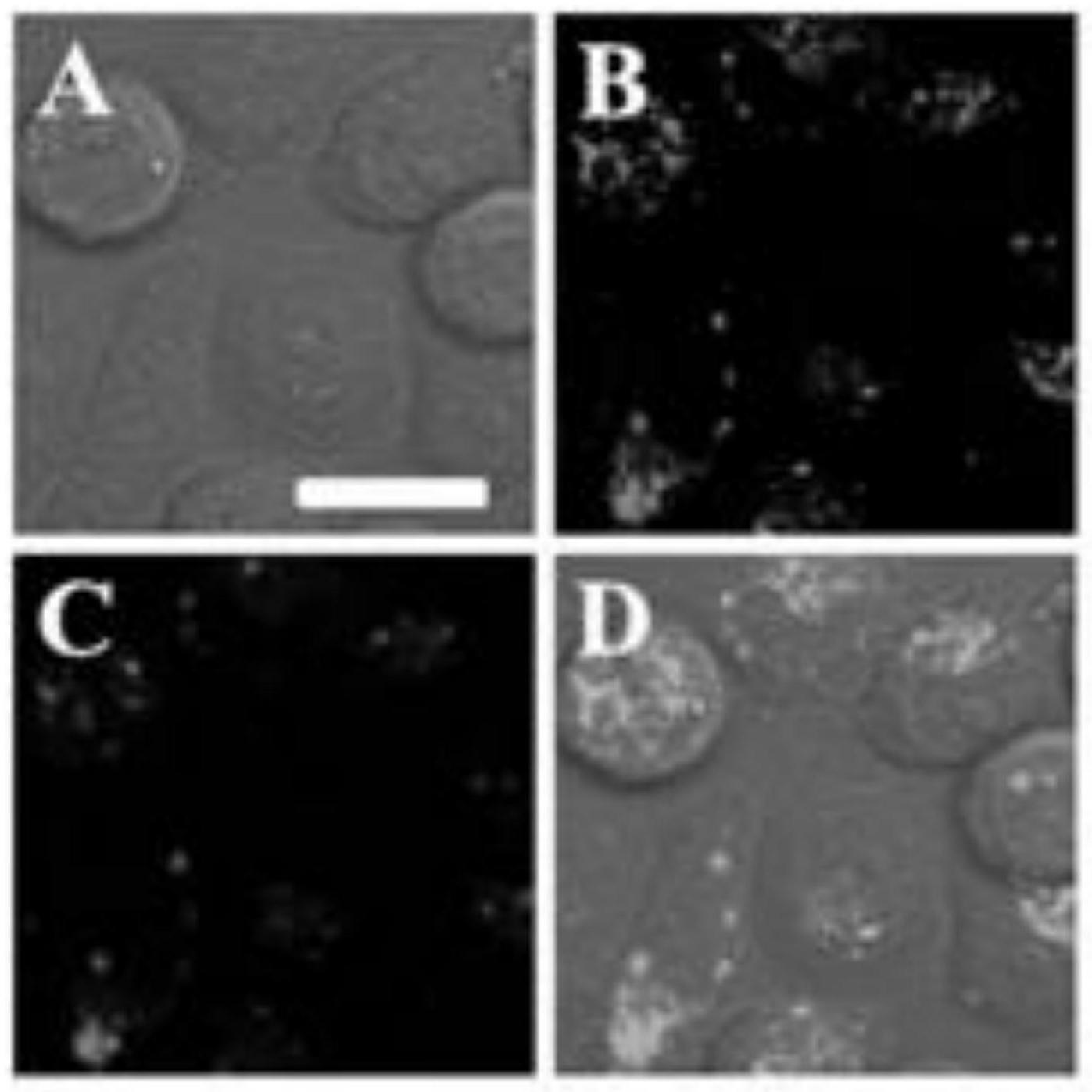
图6为聚合物p2纳米颗粒与溶酶体的共定位细胞成像。a)明场;b)溶酶体;c)聚合物p2;d)合并图像。
具体实施方式:
[0034]
实施例1
[0035][0036]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、2,6
‑
二(三甲基锡)
‑
4,8
‑
二(2
‑
辛基十二烷基氧基)
‑
苯并二噻吩(bdt
‑
od
‑
sn)(110.9mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)和cutc(0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌24小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物依次分别用丙酮、正己烷和氯仿进行索氏提取。氯仿提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到红色聚合物固体(81.6mg,84%)。1h nmr(400mhz,cdcl3):δ8.25
‑
7.50(br,2h),7.15
‑
6.73(br,2h),4.59
‑
3.59(br,4h),2.31
‑
2.06(br,2h),1.92
‑
1.12(br,64h),1.04
‑
0.64(br,12h)。gpc:m
n 15234;m
w 58645;m
w
/m
n 3.85。元素分析计算值{(c
58
h
88
n2o2s4)
n
}:c,71.70;h,8.92;n,2.88。实测值:c,70.35;h,8.64;n,2.80。该聚合物的热分解温度:328℃。最大吸收波长:520nm(氯仿溶液),520nm(薄膜)。
[0037]
实施例2
[0038][0039]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、2,7
‑
二(三甲基锡)
‑
4,4,9,9
‑
四(对己基苯)
‑
引达省并二噻吩;(idt
‑
sn)(123.3mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)和cutc(95.3mg,0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌24小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物分别用丙酮、正己烷和氯仿进行索氏提取。氯仿提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到红色聚合物(90.0mg,82%)。1h nmr(400mhz,cdcl3):δ8.43
‑
8.27(br,2h),7.62
‑
7.51(br,4h),7.23
‑
7.09(br,16h),2.64
‑
2.53(br,8h),1.66
‑
1.56(br,8h),1.36
‑
1.26(br,24h),0.92
‑
0.84(br,12h).gpc:m
n 13430;m
w 44206;m
w
/m
n 3.29。元素分析计算值{(c
72
h
74
n2s4)
n
}:c,78.93;h,6.81;n,2.56。实测值:c,76.71;h,6.89;n,2.35。该聚合物的热分解温度:416℃。最大吸收波长:536nm(氯仿溶液),539nm(薄膜)。
[0040]
实施例3
[0041][0042]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、(4,4,9,9
‑
四(4
‑
异辛基噻吩基)
‑
4,9
‑
二氢
‑
s
‑
苯并二茚并[1,2
‑
b5,6
‑
b']二噻吩
‑
2,7
‑
二基)双3,9
‑
双三甲基锡
‑
5,5,11,11
‑
四
‑
(5
‑
(2
‑
乙基己基噻吩)
‑2‑
基))
‑
噻吩[3,2
‑
b]并噻吩引达省二噻吩(三甲基锡烷)(idty
‑
sn)(148.2mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)和cutc(95.3mg,0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌24小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物分别用丙酮、正己烷、氯仿和氯苯进行索氏提取。氯仿提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到黑色聚合物(87.4mg,65%)。1h nmr(400mhz,cdcl3):δ8.48
‑
8.34(br,2h),7.97
‑
7.88(br,2h),7.83
‑
7.72(br,2h),6.90
‑
6.76(br,4h),6.64
‑
6.51(br,4h),2.76
‑
2.61(br,8h),1.66
‑
1.56(br,4h),1.34
‑
1.19(br,32h),0.92
‑
0.77(br,24h)。gpc:m
n 16404;m
w 64614;m
w
/m
n 3.94。元素分析计算值{(c
76
h
82
n2s
10
)
n
}:c,67.91;h,6.15;n,2.08。实测值:c,66.60;h,6.21;n,2.12。
[0043]
氯苯提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过
滤、烘干即可得到黑色聚合物(29.6mg,22%)。1h nmr(400mhz,cd5cl):δ8.37
‑
8.26(br,1h),8.19
‑
8.09(br,1h),7.45
‑
7.32(br,2h),7.06
‑
6.92(br,8h),6.77
‑
6.68(br,1h),6.58
‑
6.48(br,1h),2.69
‑
2.36(br,8h),1.49
‑
1.41(br,4h),1.29
‑
1.09(br,32h),0.88
‑
0.64(br,24h)。gpc:m
n 34701;m
w 109152;m
w
/m
n 3.15。元素分析计算值{(c
76
h
82
n2s
10
)
n
}:c,67.91;h,6.15;n,2.08。实测值:c,63.48;h,6.04;n,1.79。该聚合物的热分解温度:393℃。最大吸收波长:538nm(氯仿溶液),545nm(薄膜)。
[0044]
实施例4
[0045][0046]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、(4,4”'
‑
二(2
‑
己基癸基)
‑
2,,2”'
‑
二(三甲锡烷基)
‑
2,2':5',2”:5”,2”'
‑
四联噻吩(tth
‑
hd
‑
sn)(110.5mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)和cutc(95.3mg,0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌24小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物分别用丙酮、正己烷、氯仿和氯苯进行索氏提取。氯苯提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到亮黑色聚合物(58.2mg,60%)。1h nmr(400mhz,cd5cl):δ8.41
‑
7.75(br,4h),7.75
‑
7.21(br,4h),3.10
‑
2.64(br,4h),2.59
‑
2.40(br,2h),1.59
‑
1.17(br,48h),0.99
‑
0.78(br,12h)。gpc:m
n 30459;m
w 64835;m
w
/m
n 2.13。元素分析计算值{(c
56
h
74
n2s6)
n
}:c,69.52;h,7.71;n,2.90。实测值:c,66.71;h,7.55;n,2.64。该聚合物的热分解温度:399℃。最大吸收波长:462nm(氯仿溶液),498nm(薄膜)。
[0047]
实施例5
[0048][0049]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、(4,4”'
‑
二(2
‑
辛基十二基)
‑
2,,2”'
‑
二(三甲锡烷基)
‑
2,2':5',2”:5”,2”'
‑
四联噻吩(tth
‑
od
‑
sn)(121.7mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)和cutc(95.3mg,0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌24小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物分别用丙酮、正己烷、氯仿和氯苯进行索氏提取。氯仿提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到亮黑色聚合物(16.2mg,15%)。1h nmr(400mhz,cdcl3):δ8.47
‑
7.87(br,4h),7.62
‑
7.36(br,2h),7.19
‑
7.10(br,2h),2.81
‑
2.70(br,4h),1.81
‑
1.76(br,2h),1.29
‑
1.23(br,64h),0.89
‑
0.84(br,12h).。gpc:m
n 13135;m
w 52181;m
w
/m
n 3.97。元素分析计算值{(c
64
h
90
n2s6)
n
}:c,71.19;h,8.40;n,2.59。实测值:c,69.38;h,8.04;n,2.72。
[0050]
氯苯提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到亮黑色聚合物(89.8mg,83%)。1h nmr(400mhz,cd5cl):δ8.67
‑
7.26(br,8h),2.91
‑
2.78(br,2h),2.56
‑
2.47(br,2h),1.93
‑
1.88(br,2h),1.55
‑
1.21(br,64h),0.98
‑
0.83(br,12h),1.29
‑
1.09(br,32h),0.88
‑
0.64(br,24h)。gpc:m
n 45030;m
w 150865;m
w
/m
n 3.35。元素分析计算值{(c
64
h
90
n2s6)
n
}:c,71.19;h,8.40;n,2.59。实测值:c,68.09;h,8.52;n,3.02。该聚合物的热分解温度:415℃。最大吸收波长:481nm(氯仿溶液),535nm(薄膜)。
[0051]
实施例6
[0052][0053]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、2,5
‑
二(3
‑
(2
‑
正辛基十二烷基)
‑5‑
(三甲锡烷基)
‑2‑
噻吩基)噻吩并[3,2
‑
b]噻吩(ttth
‑
od
‑
sn)(119.1mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)和cutc(95.3mg,0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌24小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物分别用丙酮、正己烷、氯仿和氯苯进行索氏提取。氯苯提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到亮黑色聚合物(52.8mg,50%)。1h nmr(400mhz,cdcl3):δ8.68
‑
8.12(br,2h),7.82
‑
7.35(br,4h),3.09
‑
2.39(br,4h),2.09
‑
1.82(br,2h),1.70
‑
1.10(br,64h),0.97
‑
0.76(br,12h)。gpc:m
n 18472;m
w 44856;m
w
/m
n 2.43。元素分析计算值{(c
62
h
88
n2s6)
n
}:c,70.67;h,8.42;n,2.66。实测值:c,68.50;h,8.23;n,2.44。该聚合物的热分解温度:418℃。最大吸收波长:473nm(氯仿溶液),501nm(薄膜)。
[0054]
实施例7
[0055][0056]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、(e)
‑
1,2
‑
双(5
‑
三甲基锡
‑3‑
十四烷基噻吩
‑2‑
基)
‑
乙烯(te
‑
sn)(91.1mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)和cutc(95.3mg,0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌24小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物分别用丙酮、正己烷、氯仿和氯苯进行索氏提取。氯仿提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到黑色聚合物(46.6mg,60%)。1h nmr(400mhz,cdcl3):δ8.32
‑
7.85(br,4h),7.20
‑
6.73(br,2h),3.86
‑
3.26(br,4h),2.86
‑
2.59(br,4h),1.73
‑
1.20(br,44h),0.92
‑
0.79(br,6h)。gpc:m
n 9241;m
w 15443;m
w
/m
n 1.67。元素分析计算值{(c
46
h
64
n2s4)
n
}:c,71.45;h,8.34;n,3.62。实测值:c,73.29;h,7.64;n,1.52。
[0057]
氯苯提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到黑色聚合物(13.2mg,17%)。1h nmr(400mhz,cd5cl):δ8.85
‑
7.66(br,4h),7.00
‑
6.56(br,2h),3.98
‑
2.97(br,4h),2.82
‑
2.40(br,4h),1.88
‑
1.15(br,44h),1.07
‑
0.77(br,6h)。gpc:m
n 12871;m
w 21599;m
w
/m
n 1.68。元素分析计算值{(c
46
h
64
n2s4)
n
}:c,71.45;h,8.34;n,3.62。实测值:c,69.86;h,7.91;n,2.37。该聚合物的热分解温度:384℃。最大吸收波长:540nm(氯仿溶液),545nm(薄膜)。
[0058]
实施例8
[0059][0060]
在氮气保护下,将2,6
‑
二甲硫基苯并[1,2
‑
d:4,5
‑
d']二噻唑(28.4mg,0.1mmol)、双辛基十二烷基
‑
双三甲基锡
‑
吡咯并吡咯二酮(dpp
‑
sn)(118.7mg,0.1mmol)、pd(pph3)4(11.6mg,0.01mmol)、zn(oac)2(1.8mg,0.01mmol)和cutc(95.3mg,0.5mmol)放入反应瓶中。加入4.0ml甲苯后,在室温条件下搅拌72小时。加入20.0ml甲醇后,将生成的沉淀过滤。然后将沉淀出来的聚合物分别用丙酮、正己烷和氯仿进行索氏提取。氯仿提取的部分用氨水洗过之后,将溶液浓缩,然后用20.0ml甲醇沉淀出来,过滤、烘干即可得到蓝黑色聚合物(95.8mg,91%)。1h nmr(400mhz,cdcl3):δ9.48
‑
7.45(br,4h),7.22
‑
6.80(br,2h),4.28
‑
3.49(br,4h),2.06
‑
1.98(br,2h),1.41
‑
1.10(br,64h),0.93
‑
0.77(br,12h)。gpc:m
n 11870;m
w 53644;m
w
/m
n 4.52。元素分析计算值{(c
62
h
88
n4o2s4)
n
}:c,70.95;h,8.45;n,5.34。实测值:c,67.25;h,8.23;n,4.89。该聚合物的热分解温度:373℃。最大吸收波长:743nm(氯仿溶液),748nm(薄膜)。
[0061]
有机场效应晶体管(otfts)的制备及测试
[0062]
采用顶栅底接触器件结构,ots处理过的高p
‑
型掺杂的硅和热生长的二氧化硅(300nm)作为栅电极和栅极绝缘层。300nm sio2栅极绝缘层的电容为10nf/cm2。在用ots进行底物处理之前,晶片(在两个烧杯,分别为15分钟)在乙醇中进行溶剂超声清洗,然后用一个经过过滤的n2的流干燥,然后是20分钟的等离子清洗。将实例4和实例5所得到的聚合物分别配成20mg
·
ml
‑1的聚合物邻二氯苯溶液旋涂在ots表面成膜。在半导体膜沉积后,薄膜在真空中退火为30分钟。otfts顶部触点是将金真空蒸镀到电极上(~10
‑5torr,0.1a/s,~40nm厚),获得1400μm和40μm的通道宽度和长度。
[0063]
细胞成像(以实例2中的材料p2为共定位材料)
[0064]
a.p2纳米粒子的制备
[0065]
5mg p2(mn=13.4kda)和50mg pf127完全溶解在1ml四氢呋喃中。四氢呋喃溶液用超声波细胞粉碎机(160w)超声5分钟。超声处理之后,去离子水(5ml)一滴一滴地添加到四氢呋喃溶液中,同时超声处理10分钟。获得的纳米粒子混合溶液在室温下搅拌48小时完全除去四氢呋喃。将纳米粒子溶液在去离子水中透析2天,以去除多余的乳化剂。透析袋的截留分子量为3.5kda。得到的纳米粒子水溶液放入冰箱中以备将来使用。
[0066]
b.细胞培养
[0067]
选择hela细胞进行细胞成像。hela细胞由中国科学院大学材料科学与光电技术学院功能材料实验室提供。hela细胞使用90%的prmi
‑
1640培养基、10%的胎牛血清和1%的青霉素和链霉素的完全培养基,在37℃、5%co2细胞培养箱中培养。
[0068]
c.细胞毒性
[0069]
hela细胞被完全prmi
‑
1640培养基培养24小时,然后以10000细胞/孔的密度(n=6)接种在96孔细胞培养板。24小时后,hela细胞用不同浓度的p2培养纳米粒子(5μg
·
ml
‑1、10μg
·
ml
‑1、25μg
·
ml
‑1和50μg
·
ml
‑1)孵育。共培养24小时后,用pbs洗涤细胞三次以清除多余的纳米粒子。用cck
‑
8法测定p2纳米粒子对hela的细胞毒性。
[0070]
d.细胞成像
[0071]
将hela细胞以每孔100,000个细胞的密度接种在直径35mm的共聚焦培养皿中。hela细胞在roswell park memorial institute(rpmi)1640中培养24小时后,与p2纳米粒子(10μg
·
ml
‑1)共孵育。24小时后,用磷酸盐缓冲盐水洗涤hela细胞3次。然后,用绿色溶酶体探针(10nm)在37℃下染色1小时。用磷酸盐缓冲盐水洗涤3次hela细胞后,用lsm880共聚焦激光扫描显微镜进行细胞成像。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

