1.本发明涉及一种从蛞蝓中制备胆甾醇基十七酸酯的方法,属于中药材提取技术领域。
背景技术:
2.蛞蝓为腹足类腹足纲蛞蝓科动物的统称,为有肺的软体动物,因其体表布满黏液,俗称“鼻涕虫”。蛞蝓作为一种传统中药,常见的品种有覆套足襞蛞蝓、大蛞蝓、黄蛞蝓、野蛞蝓和双线粘液蛞蝓等,具有祛风定惊、清热解毒、破瘀通经等功用,主治筋脉拘挛、惊痫、喘息等症。现代药理学研究表明蛞蝓提取物具有抗宫颈癌、抗癌肺癌、止咳化痰平喘等功效。蛞蝓中含有多糖、氨基酸、甾醇类和萜类等化学成分,但总体来说对蛞蝓化学成分的研究不够详细。目前,仅有从蛞蝓中通过正己烷提取、乙酸乙酯萃取、乙酸乙酯萃取液中加入无水甲醇静置析出结晶,结晶再通过半制备液相制备分离得到胆固醇、β
‑
谷甾醇、链甾醇、菜籽甾醇、麦角甾醇和豆甾醇等6个甾醇的报道,以及从蛞蝓中通过超临界co2萃取、皂化、半制备液相制备得到limaxol a这1个萜类成分的报道。除此之外,尚未有从蛞蝓中分离得到别的化学成分的报道。
3.胆甾醇基十七酸酯,分子式c
44
h
78
o2,分子量638,化学结构式如下所示:
[0004][0005]
胆甾醇类成分具有防治前列腺疾病、抗乳腺癌、抗胃癌、抗肠癌、调节生长和抗病毒等功效,临床主要用于心脑血管、冠心病、前列腺肥大、胃癌、肠癌和肺癌等各种疾病的治疗。
[0006]
目前,从植物中提取分离胆甾醇基十七酸酯的方法不多,从动物中提取分离胆甾醇基十七酸酯的方法更是没有,这极大地限制了胆甾醇基十七酸酯在保健及药学领域的应用。
[0007]
鉴于此,有必要提供一种从蛞蝓中制备胆甾醇基十七酸酯的方法,以解决现有技术的不足,以及开创其新的原料来源。
技术实现要素:
[0008]
本发明的目的是克服现有技术的不足,提供一种新原料来源从源蛞蝓中制备胆甾醇基十七酸酯的方法。
[0009]
本发明解决上述技术问题的技术方案如下:一种从蛞蝓中制备胆甾醇基十七酸酯的方法,包括如下步骤:
[0010]
步骤1:正己烷提取物的制备
[0011]
称取500g粉碎后的干蛞蝓,加入6
‑
8倍原药材重量的正己烷,回流提取2
‑
4次,每次3
‑
6h,滤过,合并滤液,减压回收正己烷,得到正己烷提取物;
[0012]
步骤2:石油醚浸膏的制备
[0013]
步骤1得到的正己烷提取物,加入石油醚萃取2
‑
4次,每次300
‑
600ml,合并石油醚萃取液并浓缩,得到石油醚浸膏;
[0014]
步骤3:胆甾醇基十七酸酯流份的制备
[0015]
步骤2得到的石油醚浸膏,加入1
‑
2倍石油醚浸膏重量的二氯甲烷
‑
甲醇体系溶解,经凝胶柱层析分离,用二氯甲烷
‑
甲醇体系等梯度洗脱,分段收集流份,经薄层色谱检测,收集胆甾醇基十七酸酯流份;
[0016]
步骤4:胆甾醇基十七酸酯的制备
[0017]
步骤3得到的胆甾醇基十七酸酯流份,经减压浓缩至有晶体析出,室温下静置,过滤,干燥后,即得到胆甾醇基十七酸酯。
[0018]
本发明从蛞蝓中制备胆甾醇基十七酸酯的方法的有益效果是:
[0019]
1、本发明首次以动物蛞蝓为原料,以可重复再生利用的凝胶柱进行层析分离,以易控的二氯甲烷
‑
甲醇两相体系进行等梯度洗脱,以及结晶等方法制备胆甾醇基十七酸酯,开创了胆甾醇基十七酸酯新的原料来源。
[0020]
2、本发明的制备方法简单易操作,工艺周期短,成本低廉,市场前景广阔,适合工业化生产。
[0021]
在上述技术方案的基础上,本发明还可以做如下改进。
[0022]
进一步,步骤1中,所述粉碎后的干蛞蝓的粒径为50
‑
100目。
[0023]
采用上述进一步的有益效果是:采用上述参数,有利于后续正己烷回流提取。
[0024]
进一步,步骤2中,所述石油醚浸膏在50℃的相对密度为1.4
‑
1.7。
[0025]
采用上述进一步的有益效果是:采用上述参数,有利于后续溶解及进行柱层析分离。
[0026]
进一步,步骤3中,所述凝胶柱为sephadex lh
‑
20凝胶柱,规格2.5
×
120cm,填料粒径为27μm
‑
163μm。
[0027]
采用上述进一步的有益效果是:sephadex lh
‑
20,全称为羟丙基葡聚糖凝胶。采用上述凝胶柱层析,有利于后续胆甾醇基十七酸酯的分离纯化及重复使用节约成本与资源。上述凝胶材料来自美国ge healthcare公司。
[0028]
进一步,步骤3中,所述二氯甲烷
‑
甲醇体系中,二氯甲烷和甲醇的体积比为1:1,所述洗脱的速度为2
‑
8ml/min。
[0029]
采用上述进一步的有益效果是:采用上述参数,可以通过凝胶柱层析将胆甾醇基十七酸酯与其他成分很好的分离,且溶剂配制简单。
[0030]
进一步,步骤3中,所述薄层色谱检测的条件为:硅胶gf
254
板,展开剂为甲苯:甲酸乙酯:甲酸体积比1:7:1,直立上行展开,显色剂为质量浓度为1%的fecl3‑
乙醇溶液,显黄色单一点的洗脱部位,即为胆甾醇基十七酸酯流份。
[0031]
采用上述进一步的有益效果是:通过上述检测,可以合并收集胆甾醇基十七酸酯流份。
[0032]
上述显色剂的配制方法是:准确称量0.789g的fecl3固体,溶解于100ml无水乙醇
中,充分震荡至完全溶解,即得。
[0033]
进一步,步骤4中,所述静置的时间为8
‑
12h。
[0034]
采用上述进一步的有益效果是:有利于胆甾醇基十七酸酯充分沉淀,提高产品得率。
附图说明
[0035]
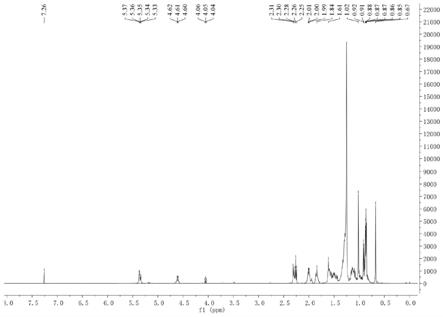
图1为本发明的实施例1中,步骤4得到的胆甾醇基十七酸酯的氢谱图(500mhz,cdcl3)。
[0036]
图2为本发明的实施例1中,步骤4得到的胆甾醇基十七酸酯的碳谱图(125mhz,cdcl3)。
具体实施方式
[0037]
以下结合具体附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
[0038]
实施例1
[0039]
本实施例的从蛞蝓中制备胆甾醇基十七酸酯的方法,包括如下步骤:
[0040]
步骤1:正己烷提取物的制备
[0041]
干黄蛞蝓粉碎至粒径为50
‑
100目,称取500g,加入8倍原药材重量的正己烷,回流提取4次,每次6h,滤过,合并滤液,减压回收正己烷,得到正己烷提取物。
[0042]
步骤2:石油醚浸膏的制备
[0043]
步骤1得到的正己烷提取物,加入石油醚萃取4次,每次600ml,合并石油醚萃取液并浓缩,得到石油醚浸膏,所述石油醚浸膏在50℃的相对密度为1.4
‑
1.7。
[0044]
步骤3:胆甾醇基十七酸酯流份的制备
[0045]
步骤2得到的石油醚浸膏,加入1
‑
2倍石油醚浸膏重量的二氯甲烷
‑
甲醇体系溶解,经sephadex lh
‑
20凝胶柱层析分离(规格2.5
×
120cm,填料粒径为27μm
‑
163μm),用二氯甲烷和甲醇的体积比为1:1的二氯甲烷
‑
甲醇体系等梯度洗脱,所述洗脱的速度为2
‑
8ml/min,分段收集流份,经薄层色谱检测,收集合并胆甾醇基十七酸酯流份。所述薄层色谱检测的条件为:硅胶gf
254
板,展开剂为甲苯:甲酸乙酯:甲酸体积比1:7:1,直立上行展开,显色剂为质量浓度为1%的fecl3‑
乙醇溶液,显黄色单一点的洗脱部位,即为胆甾醇基十七酸酯流份。
[0046]
步骤4:胆甾醇基十七酸酯的制备
[0047]
步骤3得到的胆甾醇基十七酸酯流份,经减压浓缩至有晶体析出,室温下静置12h,过滤,干燥后,即得到4.52g白色的胆甾醇基十七酸酯。
[0048]
结构鉴定:
[0049]1h
‑
nmr(500mhz,cdcl3)δ:0.67(3h,s,h
‑
18),0.86(3h,d,j=2.2hz,h
‑
27),0.87(3h,d,j=2.2hz,h
‑
26),0.92(3h,d,j=6.5hz,h
‑
21),1.02(3h,s,h
‑
19),2.25
‑
2.31(2h,m,h
‑
4),4.60
‑
4.62(1h,m,h
‑
3),5.35
‑
5.37(1h,m,h
‑
6)。氢谱图如图1所示。
[0050]
13
c
‑
nmr(125mhz,cdcl3)δ:37.2(c
‑
1),27.9(c
‑
2),73.8(c
‑
3),38.3(c
‑
4),139.9(c
‑
5),122.7(c
‑
6),32.0(c
‑
7),32.1(c
‑
8),50.2(c
‑
9),36.4(c
‑
10),21.2(c
‑
11),39.9(c
‑
12),42.5(c
‑
13),56.8(c
‑
14),24.4(c
‑
15),28.2(c
‑
16),56.3(c
‑
17),12.0(c
‑
18),19.5(c
‑
19),35.9(c
‑
20),18.9(c
‑
21),36.8(c
‑
22),22.7(c
‑
23),39.7(c
‑
24),28.4(c
‑
25),22.8(c
‑
26),23.9(c
‑
27),173.4(c
‑
1'),34.9(c
‑
2'),25.2(c
‑
3'),29.3(c
‑
4'),29.4(c
‑
5'),29.5(c
‑
6'),29.6(c
‑
7'),29.7(c
‑
8'),29.8(c
‑
9'),29.8(c
‑
10'),29.8(c
‑
11'),29.8(c
‑
12'),29.8(c
‑
13'),29.5(c
‑
14'),32.1(c
‑
15'),24.0(c
‑
16'),14.3(c
‑
17')。碳谱图如图2所示。
[0051]
通过氢谱和碳谱数据与文献报道一致,故鉴定该化合物为胆甾醇基十七酸酯。
[0052][0053]
实施例2
[0054]
步骤1:正己烷提取物的制备
[0055]
干黄蛞蝓粉碎至粒径为50
‑
100目,称取500g,加入8倍原药材重量的正己烷,回流提取4次,每次6h,滤过,合并滤液,减压回收正己烷,得到正己烷提取物。
[0056]
步骤2:石油醚浸膏的制备
[0057]
步骤1得到的正己烷提取物,加入石油醚萃取2次,每次300ml,合并石油醚萃取液并浓缩,得到石油醚浸膏,所述石油醚浸膏在50℃的相对密度为1.4
‑
1.7。
[0058]
步骤3:胆甾醇基十七酸酯流份的制备
[0059]
步骤2得到的石油醚浸膏,加入1
‑
2倍石油醚浸膏重量的二氯甲烷
‑
甲醇体系溶解,经sephadex lh
‑
20凝胶柱层析分离(规格2.5
×
120cm,填料粒径为27μm
‑
163μm),用二氯甲烷和甲醇的体积比为1:1的二氯甲烷
‑
甲醇体系等梯度洗脱,所述洗脱的速度为2
‑
8ml/min,分段收集流份,经薄层色谱检测,收集合并胆甾醇基十七酸酯流份。所述薄层色谱检测的条件为:硅胶gf
254
板,展开剂为甲苯:甲酸乙酯:甲酸体积比1:7:1,直立上行展开,显色剂为质量浓度为1%的fecl3‑
乙醇溶液,显黄色单一点的洗脱部位,即为胆甾醇基十七酸酯流份。
[0060]
步骤4:胆甾醇基十七酸酯的制备
[0061]
步骤3得到的胆甾醇基十七酸酯流份,经减压浓缩至有晶体析出,室温下静置12h,过滤,干燥后,即得到4.26g白色的胆甾醇基十七酸酯。
[0062]
实施例3
[0063]
步骤1:正己烷提取物的制备
[0064]
干黄蛞蝓粉碎至粒径为50
‑
100目,称取500g,加入8倍原药材重量的正己烷,回流提取2次,每次3h,滤过,合并滤液,减压回收正己烷,得到正己烷提取物。
[0065]
步骤2:石油醚浸膏的制备
[0066]
步骤1得到的正己烷提取物,加入石油醚萃取4次,每次600ml,合并石油醚萃取液并浓缩,得到石油醚浸膏,所述石油醚浸膏在50℃的相对密度为1.4
‑
1.7。
[0067]
步骤3:胆甾醇基十七酸酯流份的制备
[0068]
步骤2得到的石油醚浸膏,加入1
‑
2倍石油醚浸膏重量的二氯甲烷
‑
甲醇体系溶解,经sephadex lh
‑
20凝胶柱层析分离(规格2.5
×
120cm,填料粒径为27μm
‑
163μm),用二氯甲烷和甲醇的体积比为1:1的二氯甲烷
‑
甲醇体系等梯度洗脱,所述洗脱的速度为2
‑
8ml/min,分段收集流份,经薄层色谱检测,收集合并胆甾醇基十七酸酯流份。所述薄层色谱检测的条
件为:硅胶gf
254
板,展开剂为甲苯:甲酸乙酯:甲酸体积比1:7:1,直立上行展开,显色剂为质量浓度为1%的fecl3‑
乙醇溶液,显黄色单一点的洗脱部位,即为胆甾醇基十七酸酯流份。
[0069]
步骤4:胆甾醇基十七酸酯的制备
[0070]
步骤3得到的胆甾醇基十七酸酯流份,经减压浓缩至有晶体析出,室温下静置12h,过滤,干燥后,即得到4.15g白色的胆甾醇基十七酸酯。
[0071]
实施例4
[0072]
步骤1:正己烷提取物的制备
[0073]
干黄蛞蝓粉碎至粒径为50
‑
100目,称取500g,加入6倍原药材重量的正己烷,回流提取4次,每次6h,滤过,合并滤液,减压回收正己烷,得到正己烷提取物。
[0074]
步骤2:石油醚浸膏的制备
[0075]
步骤1得到的正己烷提取物,加入石油醚萃取4次,每次600ml,合并石油醚萃取液并浓缩,得到石油醚浸膏,所述石油醚浸膏在50℃的相对密度为1.4
‑
1.7。
[0076]
步骤3:胆甾醇基十七酸酯流份的制备
[0077]
步骤2得到的石油醚浸膏,加入1
‑
2倍石油醚浸膏重量的二氯甲烷
‑
甲醇体系溶解,经sephadex lh
‑
20凝胶柱层析分离(规格2.5
×
120cm,填料粒径为27μm
‑
163μm),用二氯甲烷和甲醇的体积比为1:1的二氯甲烷
‑
甲醇体系等梯度洗脱,所述洗脱的速度为2
‑
8ml/min,分段收集流份,经薄层色谱检测,收集合并胆甾醇基十七酸酯流份。所述薄层色谱检测的条件为:硅胶gf
254
板,展开剂为甲苯:甲酸乙酯:甲酸体积比1:7:1,直立上行展开,显色剂为质量浓度为1%的fecl3‑
乙醇溶液,显黄色单一点的洗脱部位,即为胆甾醇基十七酸酯流份。
[0078]
步骤4:胆甾醇基十七酸酯的制备
[0079]
步骤3得到的胆甾醇基十七酸酯流份,经减压浓缩至有晶体析出,室温下静置12h,过滤,干燥后,即得到3.99g白色的胆甾醇基十七酸酯。
[0080]
实施例5
[0081]
步骤1:正己烷提取物的制备
[0082]
干黄蛞蝓粉碎至粒径为50
‑
100目,称取500g,加入6倍原药材重量的正己烷,回流提取2次,每次3h,滤过,合并滤液,减压回收正己烷,得到正己烷提取物。
[0083]
步骤2:石油醚浸膏的制备
[0084]
步骤1得到的正己烷提取物,加入石油醚萃取2次,每次300ml,合并石油醚萃取液并浓缩,得到石油醚浸膏,所述石油醚浸膏在50℃的相对密度为1.4
‑
1.7。
[0085]
步骤3:胆甾醇基十七酸酯流份的制备
[0086]
步骤2得到的石油醚浸膏,加入2倍石油醚浸膏重量的二氯甲烷
‑
甲醇体系溶解,经sephadex lh
‑
20凝胶柱层析分离(规格2.5
×
120cm,填料粒径为27μm
‑
163μm),用二氯甲烷和甲醇的体积比为1:1的二氯甲烷
‑
甲醇体系等梯度洗脱,所述洗脱的速度为2
‑
8ml/min,分段收集流份,经薄层色谱检测,收集合并胆甾醇基十七酸酯流份。所述薄层色谱检测的条件为:硅胶gf254板,展开剂为甲苯:甲酸乙酯:甲酸体积比1:7:1,直立上行展开,显色剂为质量浓度为1%的fecl3‑
乙醇溶液,显黄色单一点的洗脱部位,即为胆甾醇基十七酸酯流份。
[0087]
步骤4:胆甾醇基十七酸酯的制备
[0088]
步骤3得到的胆甾醇基十七酸酯流份,经减压浓缩至有晶体析出,室温下静置12h,过滤,干燥后,即得到3.56g白色的胆甾醇基十七酸酯。
[0089]
对比例1
[0090]
跟实施例不同的是,对比例1的步骤2中,不是加入石油醚进行萃取,而是加入乙酸乙酯进行萃取,相应地,也没有步骤3和步骤4。对比例1的步骤2具体是:
[0091]
步骤1得到的正己烷提取物,加入石油醚萃取4次,每次600ml,合并乙酸乙酯萃取液,加入无水甲醇静置析出结晶,结晶再通过半制备液相色谱进行制备,得到2.86g白色的胆甾醇基十七酸酯。
[0092]
由此可见,用极性较大的乙酸乙酯代替石油醚进行萃取,得到的成分更复杂,很可能影响到下一步加入无水甲醇后胆甾醇基十七酸酯晶体的析出,最终导致产率偏低。
[0093]
对比例2
[0094]
跟实施例不同的是,对比例2的步骤2中,不加入石油醚进行萃取,而是加入乙酸乙酯进行萃取,得到乙酸乙酯浸膏,步骤3和步骤4不变。对比例2的步骤2具体是:
[0095]
步骤1得到的正己烷提取物,加入石油醚萃取4次,每次600ml,合并乙酸乙酯萃取液并浓缩,得到乙酸乙酯浸膏,所述乙酸乙酯浸膏在50℃的相对密度为1.4
‑
1.7。再经过步骤3的凝胶柱层析分离,分离得到的流份经薄层色谱检测时发现部分胆甾醇基十七酸酯与别的成分交叉混合严重,不能与含胆甾醇基十七酸酯主成分流份合并,最终合并含胆甾醇基十七酸酯主成流份,经减压浓缩、结晶、静置和干燥后得到3.26g白色的胆甾醇基十七酸酯。
[0096]
由此可见,用极性较大的乙酸乙酯代替石油醚进行萃取,得到的乙酸乙酯浸膏成分更复杂,影响到下一步凝胶柱层析时胆甾醇基十七酸酯的分离纯化,最终导致胆甾醇基十七酸酯的产率偏低。
[0097]
对比例3
[0098]
跟实施例不同的是,对比例3的步骤3中,凝胶柱层析分离时的洗脱剂不是用体积比为1:1的二氯甲烷
‑
甲醇体系等梯度洗脱,而是分别用体积比为4:6或6:4的二氯甲烷
‑
甲醇体系等梯度洗脱,流份经薄层色谱检测时发现部分胆甾醇基十七酸酯与别的成分交叉混合严重,不能与含胆甾醇基十七酸酯主成分流份合并,最终合并含胆甾醇基十七酸酯主成分流份,经减压浓缩、结晶、静置和干燥后分别得到3.69g及3.82g白色的胆甾醇基十七酸酯。
[0099]
由此可见,凝胶柱层析分离时的洗脱剂不用体积比为1:1的二氯甲烷
‑
甲醇体系等梯度洗脱时,不利于胆甾醇基十七酸酯的富集,最终导致胆甾醇基十七酸酯的产率偏低。
[0100]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

