4
‑
硫代脱氧胸苷衍生物及其抗乙肝病毒制药应用
技术领域
1.本发明属于药物化学领域,具体涉及一类4
‑
硫代脱氧胸苷衍生物及其抗乙肝病毒制药应用。
背景技术:
2.hbv的持续感染是造成乙肝慢性化的主要原因,且可导致病情发展、恶化、hbv相关性肝细胞癌。因此,抗病毒治疗是慢性乙肝治疗的关键。目前,常用的抗hbv药物分为免疫调节药物与核苷类似物。此外,中草药及其有效成分抗hbv治疗也占有重要地位。干扰素是hbv治疗的常见免疫调节剂,干扰素抗病毒具有广谱性,治疗效果持久并且有较高的病毒表面抗原清除率,但是其答应率低且副作用大。核苷类似物主要包括拉米夫定、替比夫定、恩替卡韦、阿德福韦、替诺福韦等,这些药物在病毒复制过程中竞争性地结合在病毒dna上,通过抑制逆转录酶活性来降低病毒dna的复制。上述药物能够减少肝功能衰竭与肝细胞癌的发生率,提高生存率。但是,这些药物有一个共同的缺点,即在使用了一段时间后将会出现耐药现象,也不能完全清除乙肝病毒。鉴于已有抗乙肝病毒药物存在的耐药性及其药效有待提高,研发新型抗乙肝病毒新型药物仍是目前治疗病毒性肝炎的重要议题。
技术实现要素:
3.发明目的:针对上述现有技术,本发明提供了一类4
‑
硫代脱氧胸苷衍生物,该4
‑
硫代脱氧胸苷衍生物包括d构型4
‑
硫代脱氧胸苷衍生物与l构型4
‑
硫代脱氧胸苷衍生物,其对hbv的抑制率和治疗率效果好,可以用于制备治疗乙型肝炎或肝癌的药物。
4.本发明还提供4
‑
硫代脱氧胸苷衍生物的应用。
5.技术方案:本发明所述的一类4
‑
硫代脱氧胸苷衍生物或其药学上可接受的盐,其结构如式i或式ii所示:;其中r1选自h,
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的烃基,甲氧基,甲硫基,
‑
c2
‑
c10的烃氧基,c2
‑
c10的烃硫基,氨甲基,羟甲基,
‑
ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2nh(ch2)nnh(ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,c3
‑
c10的脂杂环,
‑
f,
‑
cl,
‑
br,
‑
i,或氰基等;其中r2选自h,
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的烃基,甲氧基,甲硫基,
‑
c2
‑
c10的烃氧基,c2
‑
c10的烃硫基,氨甲基,羟甲基,
‑
ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2nh(ch2)nnh (ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,c3
‑
c10的脂杂环,
‑
f,
‑
cl,
‑
br,
‑
i,或氰基等;r3选自;r5选自
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的烃基,甲氧基,甲硫基,
‑
c2
‑
c10的烃氧基,c2
‑
c10的烃硫基,氨甲基,羟甲基,
‑
ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2nh(ch2)nnh (ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,或c3
‑
c10的脂杂环等;r6为,其中r8,r9单独或者同时选自h,
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的烃基,
‑
ch2ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2ch2nh(ch2)nnh (ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,或c3
‑
c10的脂杂环等,r
10
选自
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的直链烃基,
‑
c3
‑
c10的支链烃基,甲氧基,甲硫基,
‑
c2
‑
c10的烃氧基,c2
‑
c10的烃硫基,
‑
ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2ch2nh(ch2)nnh (ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,或c3
‑
c10的脂杂环等;ar选自苯基,取代苯基,c5
‑
c10的芳环,或c5
‑
c10的芳杂环等,r7具有如下结构,其中r
11
,r
12
单独或者同时选自h,
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的烃基,
‑
ch
2 ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2ch2nh(ch2)nnh (ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,或c3
‑
c10的脂杂环等,r
13
选自
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的直链烃基,
‑
c3
‑
c10的支链烃基,
‑
ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2ch
2 nh(ch2)nnh (ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,或c3
‑
c10的脂杂环等;r4选自h,
‑
ch3,
‑
ch2ch3,
‑
c3
‑
c10的烃基,甲氧基,甲硫基,
‑
c2
‑
c10的烃氧基,c2
‑
c10的烃硫基,氨甲基,羟甲基,
‑
ch2o(ch2)no(ch2)nch3(n=1
‑
3),
‑
ch2nh(ch2)nnh (ch2)nch3(n=1
‑
3),c5
‑
c10的芳环,c5
‑
c10的芳杂环,c3
‑
c10的脂环,c3
‑
c10的脂杂环,
‑
cl,
‑
br,
‑
i,或氰基等。
6.作为优选,r1选自
‑
ch3,r2选自h,r4选自h。
7.作为优选,r3选自:。
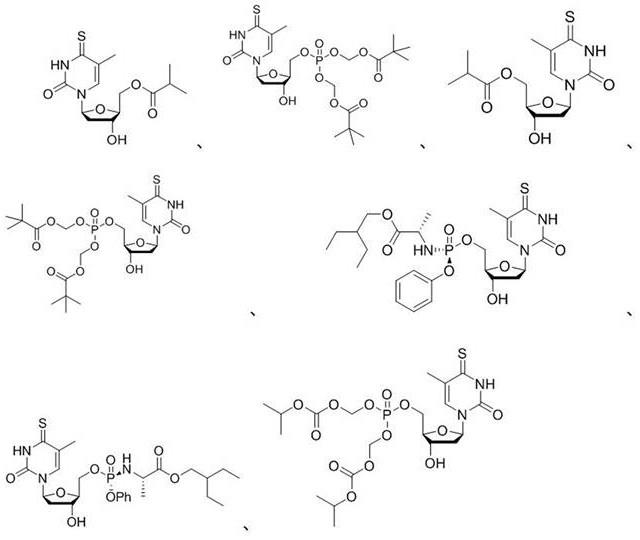
8.本发明所述衍生物或其药学上可接受的盐优选为如下任意一种:
9.作为优选,所述4
‑
硫代脱氧胸苷衍生物可作为药用盐使用,该盐包括所述化合物与金属离子或药学上可接受的胺或铵离子形成的盐。
10.本发明所述的4
‑
硫代脱氧胸苷衍生物或其药学上可接受的盐在制备治疗肝炎或肝癌的药物中的应用。
11.其中,所述肝炎为hbv感染引起的乙型肝炎。
12.本发明所述的药物组合物,包含有效量的所述的4
‑
硫代脱氧胸苷衍生物或其药学上可接受的盐、立体异构体、活性代谢物、前药、溶剂化物或结晶形式,以及药学上可以接受的赋形剂。
13.其中,所述药物组合制备成药剂学上的任意一种剂型,包括是胶囊剂、散剂、片剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂或贴剂。
14.上述4
‑
硫代脱氧胸苷衍生物、药物组合物在制备治疗乙型肝炎或肝癌的药物中的应用均在本发明的保护范围内。本发明所述乙型肝炎为hbv感染引起的。
15.有益效果:本发明提供了一组4
‑
硫代脱氧胸苷衍生物或其药学上可接受的盐,并通过体外细胞毒性试验和体外抗hbv病毒药效试验,证实4
‑
硫代脱氧胸苷衍生物或其药学上可接受的盐具有抗hbv活性,效果优于阳性对照,具有抗乙肝药物开发前景。
16.本发明4
‑
硫代脱氧胸苷衍生物或其药学上可接受的盐设计巧妙,结构简单,原料便宜易得,合成工艺安全、环保,易于规模化生产。
附图说明
17.图1是化合物q15的1hnmr谱图;图2是化合物q22的1hnmr谱图;图3是化合物q23的1hnmr谱图;图4是化合物q24的1hnmr谱图;图5是化合物q21的1hnmr谱图;图6是化合物q12的1hnmr谱图;图7是化合物q11的1hmnr图谱。
具体实施方式
18.下面通过具体实施例对本发明进行详细的说明。实施例中制备所得化合物采用代码q表示,在式i或式ii的范围内。
19.实施例1合成路线如下:
ꢀ
;胸腺嘧啶(26 g,205.7 mmol)混合于六甲基二硅氮烷(320 ml,1.48 mol)与三甲基氯硅烷(42 ml,321 mmol)中,n2保护下,130℃搅拌3h。冷却至室温后浓缩反应液,油泵抽干,向粗品中加入二氯甲烷(500 ml,无水),然后依次加入化合物a(cas:141846
‑
57
‑
3)(80 g,205.7 mmol)与三氟甲磺酸三甲基硅酯(2.25 ml,12.3 mmol),室温下搅拌1h。反应完成
后在搅拌下向反应液中缓慢加入饱和碳酸氢钠溶液,二氯甲烷萃取,有机相用饱和食盐水洗涤,分出有机相,无水硫酸钠干燥,过滤除去无水硫酸钠,滤液浓缩得到粗产品,粗产品经柱层析(石油醚:乙酸乙酯=10/1
‑
1/1)分离得到化合物b(90.3 g,黄色固体)。
20.化合物b(10.0 g,20.9 mmol)混合于甲苯(100 ml)中,n2保护下120℃搅拌直至溶液澄清,加入劳森试剂(9.3 g,23.0 mmol),80℃下搅拌2h。反应完成后冷却至室温,过滤除去不溶物,滤液浓缩,粗产品经柱层析(石油醚:乙酸乙酯=10/1
‑
1/1)分离得到化合物c(9 g,黄色固体,不纯)。
21.化合物c(20 g,41.6 mmol)溶于meoh(200ml)中,室温下加入甲醇钠的甲醇溶液(15 g,w/w=30%),室温下搅拌过夜。反应完成后冰水浴冷却,搅拌下滴加盐酸(1n)调节ph=7,浓缩反应液,经柱层析(二氯甲烷:甲醇=50/1
‑
10/1)分离得到产物d(9.66 g,黄色固体)。
[0022]1h nmr(400mhz, dmso
‑
d6)δ12.71(s,1h),7.90(s,1h),6.11(t,1h),5.28
‑
5.27(m,1h),5.10(t,1h),4.27
‑
4.22(m,1h),3.81
‑
3.79(q,1h),3.66
‑
3.54(m,2h),3.40(t,1h),3.17(d,1h),1.95(s,3h)。
[0023]
化合物d(9.66 g,40.0 mmol)溶于吡啶(150 ml)中,0℃,n2保护下加入特戊酰氯(5.25 g,43.5 mmol),室温下搅拌过夜,反应完成后浓缩反应液,粗产品经柱层析(二氯甲烷:甲醇=50/1
‑
20/1)分离得到化合物e(12.3 g,黄色固体)。
[0024]1h nmr (400 mhz, dmso
‑
d6) δ 12.77 (s, 1h), 7.54 (d, 1h), 7.54
‑
7.37(m,1h), 6.12 (t, 1h), 5.45 (d, 1h), 4.22 (d, 2h), 4.13
ꢀ–ꢀ
3.94 (m, 1h), 2.21 (dd, 2h), 1.99 (d, 3h), 1.15 (s, 9h)。
[0025]
化合物e(12.3 g,37.6 mmol)溶于二氯甲烷(200 ml,无水)中,室温下依次加入硝酸银(12.1 g,71.4 mmol)、2,4,6
‑
三甲基吡啶(38.7 g,319.4 mmol)与4
‑
甲氧基三苯基氯甲烷(34.8 g,112.7 mmol),室温下搅拌过夜。反应完成后过滤除去不溶物,滤液浓缩,粗产物经柱层析(石油醚:乙酸乙酯=10/1
‑
1/1)分离得到化合物f(16.8 g,黄色固体,不纯)。
[0026]
化合物f(16.8 g,28.0 mmol)溶于meoh(200 ml)中,室温下加入甲醇钠的甲醇溶液 (10.2g,w/w=30%),室温下搅拌过夜。反应完成后加少量水淬灭,反应液浓缩,粗产品经柱层析(石油醚:乙酸乙酯=10/1
‑
1/1)分离得到中间体化合物1(6.4 g,黄色固体)。
[0027]1h nmr (400 mhz, dmso
‑
d6) δ 12.71 (s, 1h), 7.77 (s, 1h), 7.43
‑
7.28 (m, 12h), 6.94 (d, 2h), 5.76 (s, 1h), 5.00 (s, 1h), 4.26 (s, 1h), 4.06
‑
4.01 (m, 4h), 3.18
‑
3.16 (m, 1h), 1.91 (s, 3h), 1.74
‑
1.68 (m, 2h)。
[0028]
中间体化合物1(2.4 g,4.5 mmol)与4
‑
二甲氨基吡啶(111 mg,0.9 mmol)溶于乙腈(50 ml,无水)中,室温下加入三乙胺(687 mg,6.8 mmol)和异丁酸酐(787 mg,5.0 mmol),室温下搅拌过夜,反应完成后加甲醇(10 ml),搅拌30 min,浓缩反应液,粗产物经柱层析(石油醚:乙酸乙酯=10/1
‑
1/1)分离得到产物g(2.3 g,白色固体)。
[0029]1h nmr (400 mhz, dmso
‑
d6) δ 12.75 (s, 1h), 7.47
ꢀ–ꢀ
7.24 (m, 13h), 6.97
ꢀ–ꢀ
6.90 (m, 2h), 6.11 (dd, 1h), 4.19 (dt, 1h), 4.08
ꢀ–ꢀ
3.94 (m, 1h), 3.98
ꢀ–ꢀ
3.80 (m, 2h), 3.75 (s, 3h), 2.50
ꢀ–ꢀ
2.37 (m, 1h), 1.92 (d, 3h), 1.88
ꢀ–ꢀ
1.73 (m, 2h), 0.99 (dd, 6h)。
[0030]
化合物g(2.3 g,3.8 mmol)溶于醋酸溶液(50 ml,v/v=80%)中,室温下搅拌过夜,反应完成以后,反应液浓缩,粗品经柱层析(二氯甲烷:甲醇=50/1
‑
20/1)分离得到化合物
q15(1.09 g,黄色固体)。
[0031]1hnmr谱图如图1所示,1h nmr (400 mhz, dmso
‑
d6) δ 12.76 (s, 1h), 7.56 (d, 1h), 6.12 (t, 1h), 5.45 (d, 1h), 4.24 (td, 3h), 3.96 (q, 1h), 2.56 (p, 1h), 2.30
ꢀ–ꢀ
2.14 (m, 2h), 2.01
ꢀ–ꢀ
1.96 (m, 3h), 1.09 (dd, 6h)。
[0032]
实施例2合成路线如下:
;中间体化合物1(2g,3.8 mmol)与化合物i(3.4 g,7.5 mmol,cas:1354823
‑
36
‑
1)溶于mecn(100 ml)中,加入氯化镁(359 mg,3.8 mmol),50℃下搅拌10 min,然后加入diea(1.22 g,9.4 mmol),50℃下搅拌过夜。反应结束后冷却至室温,加二氯甲烷(200 ml)稀释,柠檬酸(1n)洗涤反应液,有机相依次用饱和氯化铵洗,然后用饱和碳酸氢钠洗,饱和食盐水
洗,有机相用无水硫酸钠干燥,过滤除去无水硫酸钠,滤液浓缩,粗品经柱层析(石油醚:乙酸乙酯=10/1
‑
1/1)分离得到化合物j(2.3 g,黄色固体)。
[0033]1h nmr (400 mhz, dmso
‑
d6) δ 12.76 (s, 1h), 7.42 (d,5h), 7.36 (dd, 4h), 7.28 (ddd,6h), 7.14 (t,1h), 7.05 (d, 2h), 6.96
ꢀ–ꢀ
6.89 (m, 2h), 6.18
ꢀ–ꢀ
6.08 (m, 2h), 4.25 (d, 1h), 4.03 (q, 1h), 3.98
ꢀ–ꢀ
3.84 (m, 3h), 3.81 (dd,1h), 3.74 (s, 3h), 3.64 (ddd, 1h), 1.92 (s, 3h), 1.55 (ddd, 1h), 1.38 (hept, 1h), 1.29
ꢀ–ꢀ
1.14 (m, 8h), 0.79 (td, 6h)。
[0034]
化合物j(2.3 g,2.8 mmol)溶于醋酸溶液(80 ml,v/v=80%)中,室温下搅拌过夜,反应完成后将反应液浓缩,粗品经柱层析(二氯甲烷:甲醇=50/1
‑
20/1)分离得到化合物q23(1.4 g,黄色固体)。
[0035]1hnmr谱图如图3所示,1h nmr (400 mhz, dmso
‑
d6) δ 12.75 (s, 1h), 7.60 (d, 1h), 7.40
ꢀ–ꢀ
7.31 (m, 2h), 7.27
ꢀ–ꢀ
7.11 (m, 3h), 6.19
ꢀ–ꢀ
6.08 (m, 2h), 5.44 (d,1h), 4.24 (dq,1h), 4.22
ꢀ–ꢀ
4.09 (m, 2h), 4.01
ꢀ–ꢀ
3.94 (m, 2h), 3.93
ꢀ–ꢀ
3.77 (m, 2h), 2.13 (ddd,1h), 2.08
ꢀ–ꢀ
1.92 (m, 4h), 1.43 (hept,1h), 1.32
ꢀ–ꢀ
1.18 (m, 7h), 0.81 (t,6h)。
[0036]
实施例3合成路线如下:
;特戊酸氯甲酯(48.2 g,320 mmol)、磷酸三甲酯(11.2 g,80 mmol)与碘化钠(36 g,240 mmol)混合于乙腈(100 ml,无水),向反应液中加入几粒4a 分子筛,n2保护下90 o
c回流过夜,反应完成后反应液冷却至室温,硅藻土过滤,乙酸乙酯洗涤滤渣,滤液浓缩,粗品经柱层析(石油醚:乙酸乙酯=10/1
‑
5/1)分离得到产物l(28.3 g,无色液体)。
[0037]
化合物l(6 g,13.6 mmol)与溴化锂(1.2 g,13.6 mmol)溶于乙腈(50 ml,无水)中,90 o
c回流搅拌过夜。反应结束后冷却至室温,过滤,石油醚洗涤滤饼,滤饼经油泵拉干即为所得化合物m(4 g,白色固体)。
[0038]1h nmr (400 mhz, dmso
‑
d6) δ 5.36 (s, 2h), 5.33 (s, 2h), 1.14 (s, 18h)。
[0039]
中间体化合物1(1.9 g,3.7 mmol)与化合物m(2.4 g,7.3 mmol)溶于thf(50 ml)中,0℃下依次加入3
‑
硝基
‑
1,2,4
‑
三氮唑(834 mg,7.3 mmol),二异丙基乙胺(1.9 g,14.6 mmol)与双(2
‑
氧代
‑3‑
恶唑烷基)次磷酰氯(bop
‑
cl)(1.9 g,7.3 mmol),室温下搅拌1h。反应液加二氯甲烷(100 ml)稀释,然后加1m的柠檬酸洗涤,分出有机相,有机相加水洗涤,然后饱和食盐水洗涤。有机相用无水硫酸钠干燥,过滤,滤液浓缩,柱层析(石油醚:乙酸乙酯=10/1
‑
5/1)分离得化合物n(1.9 g,黄色固体)。
[0040]1h nmr (400 mhz, dmso
‑
d6) δ 12.75 (s, 1h), 7.49
ꢀ–ꢀ
7.40 (m, 5h), 7.36 (td,4h), 7.32
ꢀ–ꢀ
7.24 (m, 4h), 6.93 (d, 2h), 6.14 (s, 1h), 5.58
ꢀ–ꢀ
5.47 (m, 4h), 4.22 (q,1h), 3.97 (d, 1h), 3.91
ꢀ–ꢀ
3.82 (m, 1h), 3.79 (q, 1h), 3.75 (s, 3h), 1.91 (s, 3h), 1.75 (dd, 2h), 1.12 (d, 18h)。
[0041]
化合物n(1.9 g,2.2 mmol)溶于醋酸溶液(80 ml,v/v=80%)中,室温下搅拌过夜,反应完成以后,反应液浓缩,粗品经柱层析(石油醚:乙酸乙酯=10/1
‑
5/1)分离得到化合物q22(949 mg,黄色固体)。
[0042]1hnmr谱图如图2所示,1h nmr (400 mhz, dmso
‑
d6) δ 12.74 (s, 1h), 7.60 (d,1h), 6.14 (t, 1h), 5.61 (dd, 4h), 5.48 (d, 1h), 4.32
ꢀ–ꢀ
4.13 (m, 3h), 3.96 (dtd, 1h), 2.26
ꢀ–ꢀ
2.13 (m, 2h), 1.98 (d, 3h), 1.16 (d, 18h)。
[0043]
实施例4合成路线如下:
;室温下,化合物p(cas:50
‑
89
‑
5)(20.0 g,82.6 mmol)与醋酐(18.5 g,181.2 mmol)混合于吡啶(150 ml)中,室温下搅拌过夜。反应液浓缩得粗品,粗品加乙酸乙酯(200 ml)稀释,然后加柠檬酸溶液(1m)洗涤两次,水相用乙酸乙酯萃取两次,合并有机相,饱和食盐水洗涤有机相,有机相加无水硫酸钠干燥,过滤除去无水硫酸钠,有机相浓缩得到化合物
q(26.5 g,白色固体),化合物q不做进一步处理直接进行下一步反应。
[0044]1h nmr (400 mhz, dmso
‑
d6) δ 11.40 (s, 1h), 7.51 (s, 1h), 6.18 (t, 1h), 5.19 (t, 1h), 4.25
‑
4.24 (m, 2h), 4.16
‑
4.14 (m, 1h), 2.45
‑
2.42 (m, 1h), 2.30
‑
2.26 (m, 1h), 2.07 (s, 6h), 1,80 (s, 3h)。
[0045]
室温下化合物q(26.5 g,81.2 mmol)混合于甲苯(700 ml)中,97 ℃下搅拌使溶液澄清,加入劳森试剂(19.7 g,48.7 mmol),97 ℃下搅拌4h,反应液冷却至室温,浓缩反应液,柱层析(石油醚:乙酸乙酯=10/1
‑
1/1)分离得到化合物r(25.9 g,黄色固体)。
[0046]
化合物r(25.9g,75.7 mmol)溶于氨甲醇溶液(300 ml)中,室温下搅拌过夜,反应液浓缩,柱层析(二氯甲烷:甲醇=50/1
‑
10/1)分离得到中间体化合物2(22.6 g,黄色固体)。
[0047]1h nmr (400 mhz, dmso
‑
d6) δ 12.72 (s, 1h), 7.90 (s, 1h), 6.11 (t, 1h), 5.27 (d, 1h), 5.11 (t, 1h), 4,27
‑
4.25 (m, 1h), 3.81
‑
3.79 (m, 1h), 3.66
‑
3.54 (m, 2h), 2.14 (t, 2h), 1.97 (s, 3h)。
[0048]
中间体化合物2(3g,11.6 mmol)、4
‑
二甲氨基吡啶(285 mg,2.5 mmol)溶于乙腈(100 ml)中,0℃下加入异丁酸酐(2.38 g,15.1 mmol)和三乙胺(2.35 g,23.2 mmol),室温下搅拌1h。反应液加甲醇后搅拌30min。反应液浓缩,柱层析(二氯甲烷:甲醇=50/1
‑
20/1)分离得到产物q12(1.12g,黄色固体)。
[0049]1hnmr谱图如图6所示,1h nmr (400 mhz, dmso
‑
d6) δ 12.76 (s, 1h), 7.56 (s, 1h), 6.12 (t, 1h), 5,44 (d, 1h), 4.27
‑
4.22 (m, 3h), 3.98
‑
3.95 (m, 1h), 2.60
‑
2.51 (m, 1h), 2.26
‑
2.19 (m, 2h), 1.99 (s, 3h), 1.08 (d, 6h)。
[0050]
实施例5合成路线如下:;中间体化合物2(3 g,11.6 mmol)与化合物m(4 g,12.6 mmol)溶于thf(100 ml)中,0℃下依次加入3
‑
硝基
‑
1,2,4
‑
三氮唑(2.65 g,23.2 mmol)、二异丙基乙胺(6 g,46.5 mmol)和双(2
‑
氧代
‑3‑
恶唑烷基)次磷酰氯(6 g,23.2 mmol),室温下搅拌3h。反应液加乙酸乙酯(100 ml)稀释,柠檬酸(1 m)洗涤,有机相饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤除去无水硫酸钠,溶液浓缩得到粗品,粗品柱层析(石油醚:乙酸乙酯=10/1
‑
5/1)分离得到产物q11(677 mg,黄色固体)。
[0051]1hnmr谱图如图7所示,1h nmr (400 mhz, dmso
‑
d6) δ 12.76 (s, 1h), 7.60 (s, 1h), 6.16
‑
6.13 (t, 1h), 5.63
‑
5.60 (m, 4h), 5.49 (s, 1h), 4.28
‑
4.16 (m, 3h), 3.98
‑
3.95 (m, 1h), 2.20
‑
2.17 (m, 2h), 1.98 (s, 3h), 1.15 (s, 18h)。
[0052]
实施例6合成路线如下:;中间体化合物2(4 g,15.5 mmol)、化合物i (7.32 g,16 mmol)与mgcl
2 (1.62 g,17 mmol)溶于乙腈(150 ml)中,室温下50℃下搅拌30min,加入二异丙基乙胺(5g,38 mmol),50℃下搅拌过夜。反应液加乙酸乙酯(200 ml)稀释,柠檬酸(1m)洗涤两次,有机相饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤除去无水硫酸钠,滤液浓缩,柱层析(二氯甲烷:甲醇=50/1
‑
20/1)分离得到化合物q21(680 mg,黄色固体)。
[0053]1hnmr谱图如图5所示,1h nmr (400 mhz, dmso
‑
d6) δ 12.75 (s, 1h), 7.64 (s, 1h), 7.36 (t, 2h), 7.19 (q, 3h), 6.14
‑
6.06 (m, 2h), 5.42 (d, 1h), 4.26
‑
4.20 (m, 2h), 4.14
‑
4.10 (m, 1h), 4.00
‑
3.96 (m,8h), 3.92
‑
3.82 (m,2h), 2.15
‑
2.08 (m, 2h), 1.95 (s, 3h), 1.46
‑
1.42 (m, 1h), 1.32
‑
1.18 (m, 7h), 0.81 (t,6h)。
[0054]
实施例7合成路线如下:
ꢀ
;磷酸三甲酯 (20.0 g, 0.14 mol)、氯甲基异丙基碳酸酯(87.14 g, 0.57 mol)与碘化钠(64.2 g, 0.43 mol)混合于乙腈(200 ml)中,n2保护下90℃搅拌过夜,反应完成后冷却至室温,硅藻土过滤除去不溶物,滤液浓缩,粗品经柱层析(石油醚:乙酸乙酯=10/1
‑
5/1)分离得到化合物w (46.8 g,无色液体,不纯)。
[0055]
化合物w(20 g, 44.8 mmol)溶于乙腈(180 ml)中,加入溴化锂 (3.9 g, 44.8 mmol),n2保护下90℃搅拌过夜,反应完成后冷却至室温,反应液浓缩,油泵抽干得到化合物x(18.8 g,黄色油状物,不纯)。
[0056]
中间体化合物2 (2 g,7.7 mmol)与化合物x(3.64 g,8.13 mmol)溶于四氢呋喃 (60 ml)中, 0℃下依次加入3
‑
硝基
‑
1h
‑
1,2,4
‑
三唑(1.8 g,15.5 mmol)、二异丙基乙胺 (4 g,31.0 mmol)和双(2
‑
氧代
‑3‑
恶唑烷基)次磷酰氯(3.94 g,15.48 mmol),室温下搅拌1h,反应完成后加乙酸乙酯(100 ml)稀释,依次用柠檬酸水溶液(1m)洗涤,水洗,饱和食盐水洗,有机相用无水硫酸钠干燥,过滤,滤液浓缩,粗品经柱层析(石油醚:乙酸乙酯=10/1
‑
5/1)分离得到化合物q24 (1.12 g,黄色固体)。
[0057]1hnmr谱图如图4所示,1h nmr (400 mhz, dmso
‑
d6) δ 12.75 (s, 1h), 7.58 (d,1h), 6.15 (t, 1h), 5.61 (d,4h), 5.48 (d, 1h), 4.82 (heptd, 2h), 4.35
‑
4.13 (m, 3h), 4.01
‑
3.94 (m, 1h), 2.23
‑
2.08 (m, 2h), 2.01
‑
1.96 (m, 3h), 1.28
‑
1.20 (m, 12h)。
[0058]
实施例8体外抗hbv活性实验替比夫定 (购自上海泰坦科技股份有限公司),hepg2.2.15 细胞(由复旦大学药学院抗病毒药物研究室提供),胎牛血清(fbs)(赛默飞世尔生物化学制品有限公司出品),dmem培养基(赛默飞世尔生物化学制品有限公司),二氧化碳培养箱(赛默飞世尔生物化学制品有限公司),荧光定量pcr(赛默飞世尔生物化学制品有限公司)。
[0059]
本发明实施例制备的试验药品作体外抗hbv病毒药效评价试验,步骤如下:药物抗病毒活性检测:取生长良好的hepg2.2.15 细胞1瓶,用胰酶消化后,制成单细胞混悬液,用细胞计数板计数,用含10% fbs血清的dmem培养基,调整细胞密度到 2
ꢀ×ꢀ
10
5 个/ml ,接种于培养板(96孔,每孔 100ul )。置于二氧化碳培养箱,于5%co2,37℃培养至80%接触抑制。吸去上清,加入试验药物培养液,同时加入含相应浓度阳性对照药品替比夫定培养液,另外设细胞空白对照孔。每个浓度梯度和细胞空白对照设置3个复孔。置于二氧化碳培养箱,于37℃培养3d/6d/9d,吸取上清液,离心、采用荧光定量pcr 法检测上清中 hbv
‑
dna含量,结果见表 1。
[0060]
表 1
从表1中可以看出化合物q15与化合物q22对hbv的抑制率与阳性对照药替比夫定相当或者优于替比夫定,于是对化合物q15与化合物q22等化合物进行了完整的体外抗hbv实验。
[0061]
实施例9完整的体外抗hbv活性实验
本发明实施例制备的试验药品作体外细胞毒性试验和体外抗hbv病毒药效评价试验。步骤如下:(a)细胞毒性试验,取生长良好的hepg2.2.15 细胞1瓶(不同于实施例8的批次),用胰酶消化后,制成单细胞混悬液,用细胞计数板计数,用含10% fbs血清的dmem培养基,调整细胞密度到 2
ꢀ×ꢀ
10
5 个/ml ,接种于培养板(96孔,每孔 100ul )。置于二氧化碳培养箱,于5% co2,37℃培养至80%接触抑制。吸去上清,加入含不同浓度梯度试验药物培养液,同时加入含相应浓度梯度阳性对照药品替比夫定培养液,另外设细胞空白对照孔。每个浓度梯度和细胞空白对照设置3个复孔。置于二氧化碳培养箱,于37℃培养72 h,向各孔加入10 ul的 mtt(0.5/l),继续培养4 h,随后仔细吸去上清,每孔加入150 μl dmso,振摇,使甲臜颗粒溶解,最后用酶标仪比色,以空白孔调零,测546 nm处的a值。根据吸光度值计算细胞存活率和半数有毒浓度tc
50
。
[0062]
(b)药物抗病毒活性检测:取生长良好的hepg2.2.15 细胞1瓶,用胰酶消化后,制成单细胞混悬液,用细胞计数板计数,用含10% fbs血清的dmem培养基,调整细胞密度到 2
ꢀ×ꢀ
10
5 个/ml ,接种于培养板(96孔,每孔 100ul )。置于二氧化碳培养箱,于5%co2,37℃培养至80%接触抑制。吸去上清,加入含不同浓度梯度试验药物培养液,同时加入含相应浓度梯度阳性对照药品替比夫定培养液,另外设细胞空白对照孔。每个浓度梯度和细胞空白对照设置3个复孔。置于二氧化碳培养箱,于37℃培养3d/6d/9d,吸取上清液,离心、采用荧光定量pcr 法检测上清中 hbv
‑
dna含量,结果见表 2。
[0063]
表 2
注:含下划线数据可能是检测有偏差,为了数据完整性依然列于表中。
[0064]
从表2中可以看出化合物q23与化合物q24对hbv的抑制率和治疗率均大大优于阳性对照药替比夫定,化合物q22虽然没有计算出ic
50
,但是其在所有测试浓度下对乙肝病毒的抑制率均远远高于替比夫定,从其各个浓度下对hbv的抑制率看,其抗hbv强度也优于化合物q23和化合物q24。因此,化合物q22、化合物q24与化合物q23均有望成为抗hbv的具有极大潜力的新药。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。