1.本发明涉及化学医药技术领域,更具体地说,是涉及一种法匹拉韦药物共晶及其制备方法和应用。
背景技术:
2.法匹拉韦(英文名favipiravir,cas号:259793
‑
96
‑
9)化学结构式如式1所示。
[0003][0004]
法匹拉韦由日本富山化学有限公司(toyama chemical co.,ltd.)开发的一种吡嗪类似物药物,最初被批准用于抗药性流感的治疗。其作用机制是通过抑制病毒rna聚合酶进而阻断病毒rna的合成,从而起到抗病毒的作用。同时,法匹拉韦对哺乳动物细胞内的rna合成不产生抑制作用,因此是一种相对安全有效的抗病毒药物。法匹拉韦不仅可以抑制甲型和乙型流感病毒的复制,而且该药物在禽流感的治疗中也有潜在应用希望,并且可能是对神经酰胺酶抑制剂具有抗药性的流感菌株的替代选择。研究发现法匹拉韦对hiv、sars、埃博拉病毒、西尼罗病毒、黄热病病毒、黄病毒、沙粒病毒、布尼亚病毒、甲病毒、肠道病毒和里夫特裂谷热病毒等都有一定的体内或体外抗病毒作用。法匹拉韦经口服吸收良好,生物利用度高。人单次口服400mg,生物利用度高达97.6%。法匹拉韦主要在人肝胞浆中被醛氧化酶(ao)代谢为m1,然后通过肾脏排出体外。
[0005]
共晶为单一相结晶性化合物,由两种或多种分子以一定化学计量比通过非共价键结合而成。与原料药相比,共晶在溶解度、溶出速率、生物利用度以及稳定性等方面存在优势,从而引起广泛关注。然而共晶制备过程中面临制备方法与筛选方法的匮乏,采取不同的组分制备得到的共晶亦难以预料其技术效果,制备的药物共晶也存在较高的风险。
[0006]
因此,提供一种法匹拉韦药物共晶及其制备方法和应用,对于法匹拉韦药物在医学治疗的广泛应用具有重要意义。
[0007]
公开于该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域一般技术人员所公知的现有技术。
技术实现要素:
[0008]
发明目的
[0009]
为解决上述现有技术中共晶制备存在的主要问题和缺陷,本发明的目的在于提供一法匹拉韦药物共晶及其制备方法和应用。本发明法匹拉韦药物共晶为法匹拉韦与烟酰胺
按照1:1的摩尔比形成的共晶。本发明的共晶降低了法匹拉韦在水中的当量溶解度,有助于延缓法匹拉韦的溶出速率,延长法匹拉韦的起效时间,降低法匹拉韦的毒副作用。
[0010]
解决方案
[0011]
为实现本发明目的,本发明实施例提供了以下技术方案:
[0012]
第一方面,本发明提供一种法匹拉韦药物共晶;所述法匹拉韦药物共晶为法匹拉韦与烟酰胺按照1:1的摩尔比形成的共晶。
[0013]
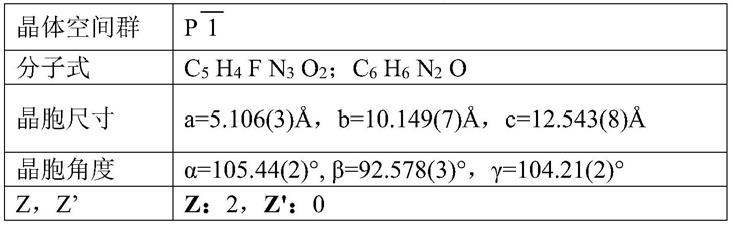
在一种可能的实现方式中,所述法匹拉韦药物共晶的晶胞属于三斜晶系,p1空间群,晶胞参数为:α=105.44(2)
°
,β=92.578(3)
°
,γ=104.21(2)
°
。本发明中法匹拉韦药物共晶内部,法匹拉韦与烟酰胺的摩尔比为1:1才能够形成共晶。
[0014]
在一种可能的实现方式中,所述法匹拉韦药物共晶以2θ角表示的x
‑
射线粉末衍射图在9.4
±
0.2
°
、14.6
±
0.2
°
、18.2
±
0.2
°
、28.2
±
0.2
°
处具有特征峰。
[0015]
在一种可能的实现方式中,所述法匹拉韦药物共晶以2θ角表示的x
‑
射线粉末衍射图还在7.1
±
0.2
°
、20.3
±
0.2
°
、22.1
±
0.2
°
、24.3
±
0.2
°
中的一处或多处具有特征峰。这是由于样品厚度、测试温度等测试因素的影响,可能会造成2θ角度的偏移。
[0016]
在一种可能的实现方式中,所述法匹拉韦药物共晶以2θ角表示的x
‑
射线粉末衍射图在28.2
±
0.2
°
处的特征峰为最强峰。
[0017]
第二方面,本发明提供一种法匹拉韦药物共晶的制备方法,所述制备方法为将法匹拉韦和烟酰胺加入溶剂中,然后冷却结晶。
[0018]
在一种可能的实现方式中,所述制备方法的具体步骤为将过量的法匹拉韦固体和烟酰胺固体加入到溶剂中,恒温下搅拌后至不能溶解,滤出清液;再按照1:1的摩尔比加入法匹拉韦与烟酰胺,加热至50~60℃,溶清后降至18~22℃恒温,过滤干燥,得到共晶。
[0019]
在一种可能的实现方式中,所述将过量的法匹拉韦固体和烟酰胺固体加入到溶剂中具体为先将过量的法匹拉韦固体加入溶剂中,恒温搅拌下法匹拉韦固体不能溶解,过滤出未溶解法匹拉韦固体;再在溶有法匹拉韦的溶剂中加入过量的烟酰胺固体,恒温搅拌下烟酰胺固体不能溶解,滤出清液。
[0020]
在一种可能的实现方式中,所述恒温搅拌的时间为6小时~12小时
[0021]
在一种可能的实现方式中,所述溶剂为乙醇。
[0022]
在一种可能的实现方式中,所述恒温温度为15~21℃。
[0023]
第三方面,本发明提供一种药物组合物,包括所述的法匹拉韦药物共晶以及药学上可接受的辅料;所述辅料包括但不限于药学上可接受的载体、稀释剂或赋形剂。
[0024]
在一种可能的实现方式中,所述药物组合物的剂型包括但不限于片剂、胶囊、注射剂、微乳剂或亚微乳剂。
[0025]
第四方面,本发明提供上述法匹拉韦药物共晶或药物组合物在制备治疗人或哺乳动物疾病的药物中的应用。
[0026]
有益效果
[0027]
相对于法匹拉韦原料药,本发明提供的法匹拉韦药物共晶中法匹拉韦在水中当量溶解度显著降低,有助于延缓法匹拉韦的溶出速率,延长法匹拉韦的起效时间,降低法匹拉韦的毒副作用。并且本发明制备共晶的组分为烟酰胺,是一种哺乳类动物必需的营养物质,
人体内无法自行合成。烟酰胺具有促进角质细胞分化、促进伤口愈合、抗衰老和抗炎等作用,并且烟酰胺毒性较低,其作为组分与法匹拉韦制备成的共晶具有良好的技术效果。
附图说明
[0028]
图1为实施例1所得法匹拉韦
‑
烟酰胺共晶的晶胞图;
[0029]
图2为实施例1所得法匹拉韦
‑
烟酰胺共晶的x射线粉末衍射图和单组分法匹拉韦以及烟酰胺的对比谱图;
[0030]
图3为实施例2所得法匹拉韦
‑
烟酰胺共晶的x射线粉末衍射图和单组分法匹拉韦以及烟酰胺的对比谱图;
[0031]
图4为实施例3所得法匹拉韦
‑
烟酰胺共晶的x射线粉末衍射图和单组分法匹拉韦以及烟酰胺的对比谱图;
[0032]
图5为实施例1所得法匹拉韦
‑
烟酰胺共晶与单组分晶体的dsc图。
具体实施方式
[0033]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0034]
另外,为了更好的说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本发明同样可以实施。在一些实施例中,对于本领域技术人员熟知的原料、方法、手段等未作详细描述,以便于凸显本发明的主旨。
[0035]
除非另有其它明确表示,否则在整个说明书和权利要求书中,术语“包括”或其变换如“包含”或“包括有”等等将被理解为包括所陈述的元件或组成部分,而并未排除其它元件或其它组成部分。
[0036]
本发明实施例中所述的室温为15℃~20℃。
[0037]
实施例1.
[0038]
一种法匹拉韦药物共晶,采用如下制备方法:称量754mg烟酰胺和972mg法匹拉韦(摩尔比1:1)混合研磨均匀,备用。在20ml乙醇中加入2g烟酰胺,室温下(15℃~20℃)搅拌6h后过滤出不溶固体,再加入0.5g法匹拉韦,搅拌6h后过滤掉不溶固体。将滤出的清液加入0.2g前述制备好的研磨过的烟酰胺和法匹拉韦混合物,加热至50℃,完全溶清。降至20℃,24小时后析出大量晶体。制备的法匹拉韦药物共晶的晶胞如图1 所示,晶胞参数如表1所示。
[0039]
表1共晶晶胞参数
[0040]
[0041]
实施例2.
[0042]
一种法匹拉韦药物共晶,采用如下制备方法:按1:1摩尔比混合烟酰胺和法匹拉韦,取0.2g混合后的烟酰胺和法匹拉韦加入2ml研磨管,再加入0.4ml乙醇,密封后用高通量珠磨机(tiss
‑
196,上海净信)以65hz的频率研磨4分钟后,可得到烟酰胺
‑
法匹拉韦共晶。
[0043]
实施例3.
[0044]
一种法匹拉韦药物共晶,采用如下制备方法:室温下称量1.5g烟酰胺溶于10ml乙醇,加法匹拉韦0.14g,加热至40~50℃溶清。再加入80mg烟酰胺以及100mg法匹拉韦,加热至50℃,溶清。室温下(15~20℃)冷却静置24小时后,析出晶体。
[0045]
实施例4.
[0046]
室温条件下(15℃~20℃),3g烟酰胺加入20ml乙醇,磁力搅拌3小时后过滤得溶有烟酰胺的乙醇饱和溶液。取溶有烟酰胺的乙醇饱和溶液10.76g,加入0.14g法匹拉韦,磁力搅拌1小时后少量固体不溶。再加0.1g烟酰胺,室温下搅拌不溶,在40℃加热后固体很快溶解,室温搅拌1h无析出。室温静置过夜后析出大颗粒单晶(100~300微米),用于单晶衍射分析。需要说明的是,实施例4所记载的单晶制备方法并不表示法匹拉韦
ꢀ‑
烟酰胺单晶的制备局限于此,利用本领域公知的单晶制备方法制得的单晶均可用于确定法匹拉韦
‑
烟酰胺的共晶的晶体结构。
[0047]
对比例1.
[0048]
按照实施例1中法匹拉韦药物共晶的制备方法,不同之处在于溶剂选择水,但是无法得到药物共晶。
[0049]
对比例2.
[0050]
按照实施例1中法匹拉韦药物共晶的制备方法,不同之处在于溶剂选择甲苯,但是无法得到药物共晶。
[0051]
对比例3.
[0052]
按照实施例1中法匹拉韦药物共晶的制备方法,不同之处在于溶剂选择乙酸乙酯,但是无法得到药物共晶。
[0053]
实验数据
[0054]
一、xrd分析
[0055]
测试方法:通过x射线粉末衍射对实施例1
‑
3所得烟酰胺与法匹拉韦的共晶进行表征;x射线粉末衍射分析采用马尔文帕纳科aeris桌面式衍射仪,x射线为cukα,测量时加入工作电压为40kv,工作电流7.5ma,扫描步长为0.02
°
,扫描速度10
°
/min,扫描范围5~40
°
。通过x射线单晶衍射对实施例4所得单晶进行表征。单晶衍射分析:在装备有turbo x射线源的布鲁克d8单晶x射线衍射仪上,采用直接驱动旋转阳极技术和cmos探测器,对实施例4制备的单晶进行了分析。使用apex3程序收集数据帧,并使用saint方法进行处理。该结构通过分析软件shelkt的本征相位法求解,并用 olex2和shelxl精修,利用全矩阵程序对所有非h原子进行各向异性精修。
[0056]
图2为实施例1所得法匹拉韦
‑
烟酰胺共晶的x射线粉末衍射图和单组分法匹拉韦以及烟酰胺的对比谱图;图3为实施例2所得法匹拉韦
‑
烟酰胺共晶的x射线粉末衍射图和单组分法匹拉韦以及烟酰胺的对比谱图;图4为实施例3所得法匹拉韦
‑
烟酰胺共晶的x射线粉末衍射图和单组分法匹拉韦以及烟酰胺的对比谱图。
[0057]
二、dsc分析
[0058]
在梅特勒
‑
托利多的dsc 3上对实施例1所得共晶进行了分析,采用一次性的普通铝坩埚,升温速度从30℃
‑
200℃,升温速度10℃/min,氮气流速10ml/min,样品在大约 120℃开始熔化。法匹拉韦和烟酰胺分别在130℃和187℃开始熔化,共晶的熔点明显低于单组分的熔点。
[0059]
三、溶解度表征
[0060]
测量方法:在带夹套的100ml玻璃反应釜中进行测量,夹套中通入20℃的循环水。反应釜中加入50ml水,并加入1~2滴的吐温80水溶液帮助固体分散。反应釜中的水用磁力搅拌,转速600rpm。分批次加入少量固体,每次间隔30min,直到反应釜中有不溶的颗粒为止,溶解过程利用目测法观察。分别考察法匹拉韦单组分晶体和实施例3制备的共晶样品的溶解度。共晶在溶解过程中,会伴随着共晶的分解而析出单组分晶体的情况,取表观溶解度的最大值。所有实验结果,测量三次取平均值。测得法匹拉韦的溶解度约为5.2
±
1mg/ml,共晶的溶解度为6.6
±
1mg/ml,换算成法匹拉韦的当量溶解度为 3.7
±
0.5mg/ml,降低了约30%。
[0061]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。