本发明提供成为恩格列净的问题的稳定性及水溶解度得到改善的新的恩格列净共结晶体。
背景技术:
共结晶体(cocrystals)是指以规则的比率通过氢键与富含如O、OH、N等一样可形成氢键的官能团或者pKa差别为3以下的共晶形成物(coformer)结合而具有新的结晶结构的结晶性固体。由于这种共结晶体包含2个分子以上的化合物,因此能够以复合体形态表现。对于这种共结晶体的形成而言,由于通过2个分子以上的新的氢键来形成结晶结构,因此可增加药物的溶解度及溶解速度。因此通过改变人体内的药物的吸收率可改变生物利用度。作为用于克服难溶性的共结晶体的共晶形成物,应当选定易于溶解在水中的与水亲和的分子。如果选定与水不亲和而且溶解度差的共晶形成物,则不能克服药物的难溶性,反而会引起溶解度降低的现象。因此,共晶形成物的选定成为尤为重要的必要条件。并且,共结晶体包括无定形多晶型、水合物、溶剂化物等。结晶(crystals)大体上在结晶化过程中优先析出亚稳结晶之后,在溶剂介质及固体状态下转移为更稳定的结晶结构,由此具有要保持稳定性的特性,因此大体上在溶剂内通过相变(phasetransformation)重新排列分子来形成更稳定的结晶结构。因此,生成多晶型,这被称作奥斯瓦尔德(ostwald)的稳定态规律阶段(ruleofstage)。故而,亚稳结晶和稳定结晶的物理化学特性发生变化。因此,引起表示作为药物最为重要的结晶的热力学能的溶解度的变化。并且,共结晶体的共晶形成物大体上在溶剂内,通过由溶剂分子代替的相变现象,可确认结晶结构发生变化或者共结晶体自身的多晶型的存在。故而,共结晶体在水中成为水合物可相变,因此,可引起共结晶体的溶解度变成水合物的溶解度的现象。在难溶性水合物结晶型中,即使制备共结晶体也不能改善溶解度的理由在于,共结晶体相变为水合物。因此,选定共晶形成物尤为重要。根据使用何种共晶形成物,可抑制或促进相变(CrystalGrowth&Design(2011)11,887-895,RecrystallizationinMaterialsProcessingIntech:Vienna,Austria,2015;pp.173-74,DrugDiscoveryToday(2008)13,440-446)。通过共结晶化(cocrystallization)来形成共结晶体。共结晶化方法有溶剂蒸发法(solventevaporationtechnique)、抗溶剂法(anti-solventmethod)、溶剂滴加研磨法(solventdropgrinding)、淤浆法(slurrytechnique)、固相研磨法(solidstategrinfing)等(RecrystallizationinMaterialsProcessingIntech:Vienna,Austria,2015;pp.173-74)。无定形(amorphous)是指虽然存在分子的相互作用但不能形成结晶排列的固体状态。因此,具有比起结晶型更高的能级,由此其溶解度高于结晶型的溶解度。但是,因高的能级而导致热力学稳定性低,由此具有很快相变为结晶型的问题。无定形形态的共结晶体(co-amorphous)通过两个分子以上的分子间的新的氢键来形成无定形固体,由此可呈现抑制相变为结晶型的现象的效果,其为可通过高溶解度来克服难溶性的固体状态。像这样,可抑制相变为结晶型的现象的理由在于,共结晶体无定形形态的玻璃化转变温度高于药物自身的无定形的玻璃化转变温度(AdvancedDrugDeliveryReviews(2016)100,116-125)。并且,无定形形态的共结晶体的形成,应当使用非常快地控制结晶化速度,来快速达到高的过饱和度的结晶化方法。典型使用减压蒸发结晶化、超临界结晶化、液氮结晶化、冷冻蒸发结晶化等非常极度加快结晶化速度的方法。但是,由于在共结晶体的形成过程中,基于药物分子与共晶形成物之间的相互作用的氢键非常多样,因此即使使用这种极端的结晶化方法也很难设计及控制无定形(FromMoleculestoCrystallizersAnIntroductiontoCrystallization2000;pp.2-14)。并且,为了在无定形形态的共结晶体中确认稳定性的提高,需要通过温度差示扫描量热法(DSC)分析来确认玻璃化转变温度(Tg)是否在比起以往药物的无定形更高的温度下出现。这理由在于,可表示无定形形态可存在的温度即为玻璃化转变温度(AdvancedDrugDeliveryReviews100(2016)116-125)。SGLT-2(钠/葡萄糖共转运体2)是在肾脏与SGLT-1(钠/葡萄糖共转运体1)仪器负责过度的血糖再吸收的输送体,SGLT-2起到大部分的作用。因此,若SGLT-2抑制剂抑制SGLT-2输送体,则通过小便排出的血糖会增加,最终导致血糖降低,进而排出血糖持有的卡路里,产生减少体重的效果。开发为以这种效果可有用地使用为二型糖尿病治疗剂的SGLT-2抑制剂的药物中之一为恩格列净(Empagliflozin),由勃林格殷格翰公司开发,目前以欧唐静片的商品名销往全球各地。恩格列净由以下结构式(化学式1)表示,并揭示在国际公开专利公报WO2005/092877号中。[化学式1]在国际公开专利公报WO2006/117359号中揭示了恩格列净结晶型,在国际公开专利公报WO2011/039107号中揭示了用于制备恩格列净结晶型的结晶化方法。但是,据报道,国际公开专利公报WO2006/117359号中揭示的恩格列净结晶型的水溶解度仅达到0.11mg/mL,稳定性也低。并且,恩格列净作为原料医药品,由于稳定性低,因此存在难以保持规定质量而不能有用于制剂学的缺点。在国际公开专利公报WO2011/039107号中揭示了恩格列净结晶型的制备方法,但存在使用离心分离及冷却灯的结晶化方法的复杂性问题。因此,本发明为了克服恩格列净的低水溶解度及不能保持固定质量的稳定性,利用更容易操作而生产效率容易提高的结晶化方法,由此开发出恩格列净共结晶体。在本说明书全文中,参照了多篇论文及专利文献,并表示了其引用内容。所引用的论文及专利文献的公开内容,全部插入到本说明书中供参考,由此更加明确说明本发明所属的
技术领域:
的水准及本发明的内容。[现有技术文献][专利文献](专利文献1)国际公开专利公报WO2005/092877号(专利文献2)国际公开专利公报WO2006/117359号(专利文献3)国际公开专利公报WO2011/039107号技术实现要素:技术问题为了改善成为恩格列净的问题的低水溶解度及稳定性而努力而得出的结果,本发明人开发出恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体。因此,本发明的目的在于,提供新的恩格列净/富马酸、恩格列净/柠檬酸、恩格列净/L-焦谷氨酸共结晶体。本发明的另一目的在于,提供上述的本发明的新的恩格列净/富马酸、恩格列净/柠檬酸、恩格列净/L-焦谷氨酸共结晶体的制备方法。本发明的其他目的及优点通过以下的
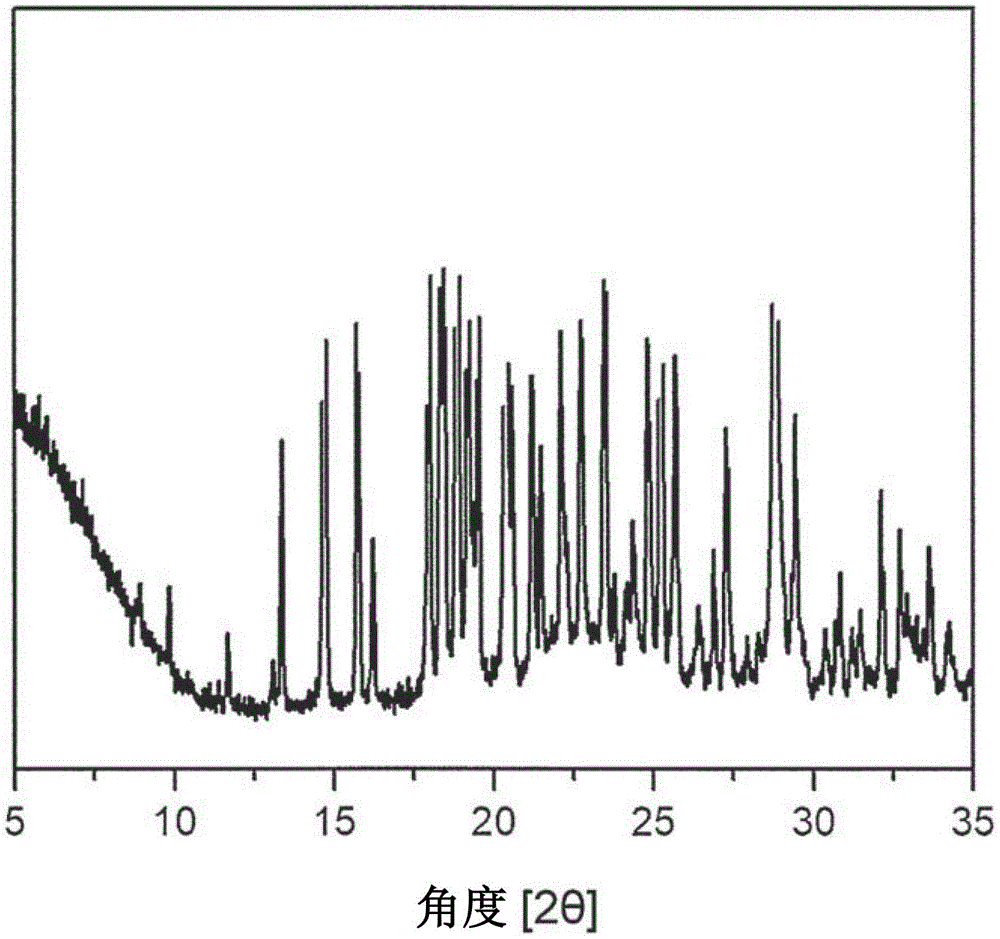
发明内容、权利要求书及附图变得更加明确。解决问题的方案根据本发明的一实施方式,本发明提供由一个分子的恩格列净和一个分子的富马酸结合而成的共结晶体。根据本发明的一实施方式,本发明提供由一个分子的恩格列净和一个分子的柠檬酸结合而成的无定形形态的共结晶体。根据本发明的一实施方式,本发明提供由一个分子的恩格列净和一个分子的L-焦谷氨酸结合而成的共结晶体。本发明人不利用碱性无机盐类,而使用作为氨基酸或有机酸的富马酸、柠檬酸、L-焦谷氨酸,来制备恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体。因此,本发明人为了克服恩格列净的低稳定性以及水溶解度,选定水溶解度非常高,且富含NH、N、O、OH的L-脯氨酸、L-精氨酸、L-赖氨酸、富马酸、草酸、L-焦谷氨酸、柠檬酸,试图设计及制备共结晶体。其结果,开发出恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体。因此,通过实验最佳化,确立了有再现性地制备恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体的方法,如此制备的恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体相比于以往恩格列净结晶型,水溶解度及小肠pH的溶解度增加约120倍,由于在摄取口服剂时投入的水量250ml中可充分溶解,因此可改善口服吸收度,也可提高吸湿性及稳定性。因此,若使用本发明的恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体,则可适当应用为改善恩格列净的稳定性及水溶解度的改良新药。根据本发明的一实例,上述恩格列净/富马酸共结晶体是由以下化学式2表示的化合物:[化学式2]对于化学式2的恩格列净/富马酸共结晶体而言,通过氢键,以1:1比率由一个分子的恩格列净和一个分子的富马酸形成共结晶体。具有如上高的水溶解度的富马酸通过氢键与恩格列净形成共结晶体,因与富马酸之间的相互作用,当富马酸溶解时,恩格列净也一起在水中溶解,因此水溶解度及小肠pH6.8溶解度增加。本发明的恩格列净/富马酸共结晶体是尚未在任何文献中报道的新型固体形态。根据本发明的实例,提供由以下化学式2表示的恩格列净/富马酸共结晶体,其特征在于,上述的恩格列净/富马酸共结晶体,在粉末X射线衍射(PXRD)分析中,具有2θ衍射角在14.703±0.2、15.747±0.2、17.958±0.2、18.859±0.2、19.192±0.2、19.518±0.2、20.367±0.2、25.23±0.2及28.794±0.2中具有特有峰值的粉末X射线衍射图案。例如,本发明的恩格列净/富马酸共结晶体的一实例的粉末X射线衍射的强度及峰值位置可如下[表1]所示。[表1](恩格列净/富马酸共结晶体的PXRD强度及峰值位置)并且,本发明提供如下的恩格列净/富马酸共结晶体,其在使用密封盘的温度差示扫描量热法(DSC)分析中,采用10℃/min的升温速度时,吸热起始温度为145.78℃±3℃,吸热温度为148.10℃±3℃,其分子的比率以1:1形成。并且,本发明提供如下的恩格列净/富马酸共结晶体,在核磁共振分光(NMR)分析中,1H-NMR光谱明确为一个分子的恩格列净和一个分子的富马酸,其分子的比率以1:1形成。根据本发明的一实例,上述无定形形态的恩格列净/柠檬酸共结晶体为由以下化学式3表示的化合物:[化学式3]对于化学式3的无定形形态的恩格列净/柠檬酸共结晶体而言,通过氢键,由一个分子的恩格列净和一个分子的柠檬酸以1:1比率形成共结晶体。具有如上高的水溶解度的柠檬酸通过氢键与恩格列净形成无定形形态,因与柠檬酸的相互作用,当柠檬酸溶解时,恩格列净也一起在水中溶解,因此水溶解度及小肠pH6.8溶解度增加。本发明的无定形形态的恩格列净/柠檬酸共结晶体是尚未在任何文献中报道的新型固体形态。根据本发明的实例,上述无定形形态的恩格列净/柠檬酸共结晶体或复合剂是其分子的比率以1:1形成的无定形形态的共结晶体或复合剂,其在粉末X射线衍射(PXRD)分析中,2θ衍射角呈现无定形形态,在温度差示扫描量热法(DSC)分析中,热量曲线呈现无定形形态。并且,本发明提供如下的无定形形态的恩格列净/柠檬酸共结晶体,在核磁共振分光(NMR)分析中,1H-NMR光谱明确为一个分子的恩格列净和一个分子的柠檬酸,其分子的比率以1:1形成。根据本发明的一实例,上述恩格列净/L-焦谷氨酸共结晶体为由以下化学式4表示的化合物:[化学式4]对于化学式4的恩格列净/L-焦谷氨酸共结晶体而言,通过氢键,由一个分子的恩格列净和一个分子的L-焦谷氨酸以1:1比率形成共结晶体。具有如上高的水溶解度的L-焦谷氨酸通过氢键与恩格列净形成共结晶体,因与L-焦谷氨酸的相互作用,当L-焦谷氨酸溶解时,恩格列净也一起在水中溶解,因此水溶解度及小肠pH6.8溶解度增加。本发明的恩格列净/L-焦谷氨酸共结晶体是尚未在任何文献中报道的新型固体形态。根据本发明的实例,提供由化学式4表示的恩格列净/L-焦谷氨酸共结晶体,其特征在于,上述的恩格列净/L-焦谷氨酸共结晶体,在粉末X射线衍射(PXRD)分析中,具有2θ衍射角在14.738±0.2、18.001±0.2、18.892±0.2、20.418±0.2、22.226±0.2、23.041±0.2、24.878±0.2、25.712±0.2及27.306±0.2中具有特有峰值的粉末X射线衍射图案。例如,本发明的恩格列净/L-焦谷氨酸共结晶体的一实例的粉末X射线衍射的强度及峰值位置可如以下[表2]所示。[表2](恩格列净/L-焦谷氨酸共结晶体的PXRD强度及峰值位置)并且,本发明提供如下的恩格列净/L-焦谷氨酸共结晶体,其在使用密封盘的温度差示扫描量热法(DSC)分析中,采用10℃/min的升温速度时,吸热起始温度为122.98℃±3℃,吸热温度为127.09℃±3℃,其分子的比率以1:1形成。并且,本发明提供如下的恩格列净/L-焦谷氨酸共结晶体,在核磁共振分光(NMR)分析中,1H-NMR光谱明确为一个分子的恩格列净和一个分子的L-焦谷氨酸,其分子的比率以1:1形成。根据本发明的另一实施方式,本发明提供包括如下步骤的恩格列净的共结晶体的制备方法。步骤(a),混合恩格列净及有机溶剂,其中分别添加有机酸;步骤(b),对上述步骤(a)的结果物进行升温,回流搅拌;步骤(c),冷却上述步骤(b)的结果物,搅拌;步骤(d),蒸发上述步骤(c)的溶剂的1/2;以及步骤(e),真空干燥上述步骤(d)的结果物,获得恩格列净共结晶体。本发明人确立了制备恩格列净的药剂学上有用的新型共结晶体,并在制备过程中即使不追加额外添加或去除盐类的工序也能够以高收率制备非常纯的新型共结晶体的方法。该方法被称作共结晶化中的溶剂蒸发法。由于本发明的方法是用来制备上述本发明的恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体,因此省略这些记载内容之间的共同内容,以便避免因重复记载导致的说明书的过度的复杂性。以下,按不同步骤,详细说明用于制备恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体的本发明的方法如下:(a)恩格列净及有机溶剂混合及有机酸的添加首先,本发明的方法包括混合固体粉末的恩格列净及有机溶剂,其中分别添加有机酸的步骤。根据本发明的一实例,上述步骤(a)的恩格列净,相对于有机溶剂的体积,添加为1-30(w/v)%,更优选地添加为6-25(w/v)%,进而优选地添加为10-20(w/v)%。制备上述恩格列净共结晶体时,被证明为以高收率生成恩格列净共结晶体,也有效去除过量使用的有机酸的溶剂的有机溶剂,优选地选自由甲醇、乙醇、异丙醇、丙酮、四氢呋喃、二甲基乙酰胺、二甲基亚砜、二甲基甲酰胺、氯仿、甲基乙基酮、乙酸乙酯、二氯甲烷及乙腈组成的有机溶剂中的1种以上,即,选择单一溶剂或其混合溶剂,更优选为甲醇、乙醇、异丙醇或丙酮,最优选为甲醇。根据本发明的另一实例,相对于恩格列净,以1至1.5当量的摩尔比添加上述步骤(a)的有机酸。优选地,相对于恩格列净,分别以1至1.5当量的摩尔比添加有机酸。如此获得的无定形形态的恩格列净新型共结晶体或复合剂成为与恩格列净结合的无定形形态的恩格列净/有机酸共结晶体或复合剂。根据本发明的另一实例,上述步骤(a)的有机酸,分为1/3的量添加到上述混合的恩格列净及有机溶剂中。这是因为,制备上述恩格列净/有机酸共结晶体或复合剂时,混合固体粉末恩格列净和有机酸时,有机酸和恩格列净的混合比率成为1:1比率尤为重要。对此,相比于一次性添加与固体粉末恩格列净混合的有机酸时,分批混合整体混合当量的1/3的有机酸时可最有效地形成。更优选地,将上述步骤(a)的有机酸分为1/3的量,隔着每20分钟,添加到上述混合的恩格列净及有机溶剂中。(b)上述结果物的升温及回流搅拌接着,本发明的方法经过对上述步骤(a)的结果物进行升温,回流搅拌的步骤。上述步骤(a)的结果物的升温时间需要1小时以上的时间,此时需要将温度调节至达到回流温度为止,在回流温度下的搅拌时间不得超过三个小时。根据本发明的另一实例,将上述步骤(b)的升温执行1小时至3小时。根据本发明的另一实例,将上述步骤(b)的回流搅拌执行1小时至3小时,更优选地执行30分钟至2小时,进而优选地执行30分钟至1小时。(c)上述结果物的冷却及搅拌接着,本发明的方法包括冷却上述步骤(b)的结果物即回流的反应液并搅拌的步骤。根据本发明的另一实例,在15-40℃下执行上述步骤(c)的冷却,更优选地在15-30℃温度下执行,最优选地在15-20℃下执行。根据本发明的另一实例,将上述步骤(c)的搅拌执行1-12小时,更优选地执行1小时至8小时,进而优选地执行3小时至5小时。(d)上述结果物的蒸发及搅拌接着,本发明的方法包括使上述步骤(c)的结果物即冷却的反应液的溶剂的1/2蒸发并搅拌的步骤。根据本发明的另一实例,在30-60℃下执行上述步骤(d)的蒸发,更优选地在30-50℃温度下执行,进而优选地在30-40℃下执行。根据本发明的另一实例,将上述步骤(d)的搅拌执行1-12小时,更优选地执行1小时至8小时,进而优选地执行3小时至5小时。根据本发明的另一实例,在上述步骤(d)之后还包括对上述步骤(d)的结果物进行减压过滤之后用有机溶剂洗涤的步骤(d-1)。根据本发明的另一实例,上述步骤(d-1)的有机溶剂选自由甲醇、乙醇、异丙醇、丙酮、四氢呋喃、二甲基乙酰胺、二甲基亚砜、二甲基甲酰胺、氯仿、甲基乙基酮、乙酸乙酯、二氯甲烷及乙腈组成的有机溶剂中的1种以上,即选择的单一溶剂或其混合溶剂,更优选为甲醇、乙醇、异丙醇或丙酮,最优选为丙酮。根据本发明的另一实例,上述步骤(c-1)的有机溶剂,相对于上述步骤(a)的有机溶剂的体积,添加为1-30(v/v)%,更优选地添加为1-10(v/v)%,进而优选地添加为2-5(v/v)%。(e)上述结果物的真空干燥及恩格列净共结晶体的获得最终,本发明的方法经过对上述步骤(e)的结果物进行真空干燥并获得恩格列净共结晶体的步骤。根据本发明的另一实例,上述步骤(e)的真空干燥在30-65℃温度下执行8小时至12小时。更优选地,在40-55℃温度下执行上述真空干燥,更优选地在45-50℃温度下执行。在上述温度及时间内执行真空干燥来获得的无定形形态的恩格列净共结晶体水分含量为1%以下,基于高效液相色谱法HPLC的纯度为99.5%以上。通过这种方法可制备由1当量的恩格列净和1当量的有机酸结合而成恩格列净共结晶体,其作为SGLT-2抑制剂在肾脏中抑制血糖的再吸收冰通过小便排出血糖,由此可用作调节血糖的二型糖尿病治疗剂。发明的效果本发明的特征及优点概略如下:(a)本发明提供由一个分子的恩格列净和一个分子的有机酸结合而成的恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体及其制备方法。(b)本发明的恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体,在制备过程中通过共结晶化的特殊溶剂环境下,调节有机酸的当量、搅拌溶剂的蒸发量、溶剂的蒸发温度及时间,能够以优秀的纯度及收率获得最佳比率的新型共结晶体。(c)对于本发明的恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体而言,借助本发明的共结晶体,将非常低的恩格列净的稳定性及水溶解度增加为如下程度,即,恩格列净的水溶解度及小肠pH6.8的溶解度增加到120倍,而且稳定性也得到提高,由此非常有用地作为恩格列净原料医药品。附图说明图1示出根据本发明的实施例制备的恩格列净/富马酸共结晶体的粉末X射线衍射(PXRD)图案结果。图2示出根据本发明的实施例制备的无定形形态的恩格列净/柠檬酸共结晶体的粉末X射线衍射(PXRD)图案结果。图3示出根据本发明的实施例制备的恩格列净/L-焦谷氨酸共结晶体的粉末X射线衍射(PXRD)图案结果。图4示出根据国际公开专利公报WO2006/117359号的实施例制备的恩格列净结晶型的粉末X射线衍射(PXRD)图案结果。图5示出根据本发明的实施例制备的恩格列净/富马酸共结晶体的温度差示扫描量热法(DSC)的热量曲线结果。图6示出根据国际公开专利公报WO2006/117359号的实施例制备的恩格列净结晶型的温度差示扫描量热法(DSC)的热量曲线结果。图7示出根据本发明的实施例制备的无定形形态的恩格列净/柠檬酸共结晶体的温度差示扫描量热法(DSC)的热量曲线结果。图8示出根据本发明的实施例制备的无定形形态的恩格列净/柠檬酸共结晶体、柠檬酸、恩格列净结晶型的温度差示扫描量热法(DSC)的热量曲线结果。图9示出根据本发明的实施例制备的恩格列净/L-焦谷氨酸共结晶体的温度差示扫描量热法(DSC)的热量曲线结果。图10示出根据本发明的实施例制备的恩格列净/L-焦谷氨酸共结晶体、L-焦谷氨酸、恩格列净结晶型的温度差示扫描量热法(DSC)的热量曲线结果。图11示出根据本发明的实施例制备的恩格列净/富马酸共结晶体的核磁共振分光(NMR)1H-NMR光谱结果,在该结果中,恩格列净/富马酸的化学计量比率准确为1:1,其峰值被积分。图12示出根据本发明的实施例制备的无定形形态的恩格列净/柠檬酸共结晶体的核磁共振分光(NMR)1H-NMR光谱结果,在该结果中,恩格列净/富马酸的化学计量比率准确为1:1,其峰值被积分。图13示出根据本发明的实施例制备的恩格列净/L-焦谷氨酸共结晶体的核磁共振分光(NMR)1H-NMR光谱结果,在该结果中,恩格列净/富马酸的化学计量比率准确为1:1,其峰值被积分。图14示出根据国际公开专利公报WO2006/117359号的实施例制备的恩格列净结晶型的核磁共振分光(NMR)1H-NMR光谱结果。图15为根据本发明的实施例制备的恩格列净/富马酸共结晶体、恩格列净/柠檬酸无定形形态的共结晶体、恩格列净/L-焦谷氨酸共结晶体以及恩格列净结晶型的1mg/mL的浓度的水溶解性的比较试验结果。此时,虽然本发明的恩格列净共结晶体均被溶解,但是恩格列净结晶型的溶解度低而不被溶解。具体实施方式以下,通过实施例更详细说明本发明。这些实施例仅用于更具体说明本发明,根据本发明的主旨,本发明的范围不限定于这些实施例,这对于本领域普通技术人员而言是显而易见的。[实施例1]恩格列净/富马酸共结晶体的制备放入恩格列净10g和甲醇100ml之后,在常温下搅拌20分钟。然后,将富马酸1.2当量各自投入1/3,将温度缓慢提高1小时,直至达到回流温度为止,然后进一步搅拌30分钟。缓慢冷却至15℃-20℃之后,搅拌3小时。然后,在30℃-40℃的温度下搅拌3小时,使甲醇70ml蒸发而析出结晶体。对析出的结晶体进行减压过滤之后,用丙酮10ml洗涤,在45℃下真空干燥16小时以上,由此获得新的恩格列净/富马酸共结晶体90%收率。[实施例2]恩格列净/柠檬酸共结晶体的制备放入恩格列净10g和甲醇100mL之后,在常温下搅拌20分钟。然后,投入柠檬酸1.2当量之后,搅拌1小时。然后,在30℃-40℃的温度下搅拌3小时,使甲醇80mL蒸发而析出结晶体。然后,投入乙酸乙酯50mL之后,搅拌20分钟。并且,对析出的结晶体进行减压过滤之后,用乙酸乙酯10ml洗涤,在45℃下真空干燥16小时以上,由此获得新的恩格列净/柠檬酸共结晶体85%收率。[实施例3]恩格列净/L-焦谷氨酸共结晶体的制备放入恩格列净10g和甲醇100ml之后,在常温下搅拌20分钟。然后,将L-焦谷氨酸1.2当量各自投入1/3,将温度缓慢提高1小时,直至达到回流温度为止,然后进一步搅拌30分钟。缓慢冷却至15℃-20℃之后,搅拌3小时。然后,在30℃-40℃的温度下搅拌3小时,使甲醇60ml蒸发而析出结晶体。对析出的结晶体进行减压过滤之后,用乙酸乙酯10ml洗涤,在45℃下真空干燥16小时以上,由此获得新的恩格列净/L-焦谷氨酸共结晶体82%收率。[实施例4]恩格列净/富马酸共结晶体的制备放入恩格列净10g和甲醇300ml之后,在常温下搅拌20分钟而溶解。然后,投入富马酸1.2当量之后,搅拌30分钟而溶解。然后,利用浓缩机,将甲醇全部蒸发掉,直至析出结晶体为止。并且,投入乙酸乙酯50mL,搅拌20分钟之后,减压过滤,用乙酸乙酯10ml洗涤,在45℃下真空干燥16小时以上,由此获得新的恩格列净/富马酸共结晶体88%收率。[实施例5]恩格列净/L-焦谷氨酸共结晶体的制备放入恩格列净10g和甲醇300ml之后,在常温下搅拌20分钟而溶解。然后,投入L-焦谷氨酸1.2当量之后,搅拌30分钟而溶解。然后,利用浓缩机,将甲醇全部蒸发掉,直至析出结晶体为止。并且,投入乙酸乙酯50mL,搅拌20分钟之后,减压过滤,用乙酸乙酯10ml洗涤,在45℃下真空干燥16小时以上,由此获得新的恩格列净/L-焦谷氨酸共结晶体83%收率。[实验例1]粉末X射线衍射(PXRD)利用CuKα放射线,在(D8Advance)X射线粉末衍射仪上执行PXRD分析(参照图1至图4)。在仪器上安装管动力装置,电流量设置为45kV及40mA。发射及散射狭缝设置为1°,受光狭缝设置为0.2mm。将2θ测定至5~35°为止,以3°/分钟(0.4秒/0.02°间隔)移动的速度进行θ-2θ连续扫描。[实验例2]温度差示扫描量热法(DSC)使用从TA公司得到的DSCQ20,在氮净化下,在20℃至300℃下,以10℃/min扫描速度,在密封盘中执行DSC测定(参照图5至图10)。[实验例3]恩格列净共结晶体的溶解度评价由于恩格列净呈具有0.111mg/ml的水溶解度的难溶性,因此与水溶解度高的富马酸、柠檬酸、L-焦谷氨酸结合而制备新的共结晶体,由此改善恩格列净的水溶解度及胃肠道pH溶解度。其结果整理在以下表3中。[表3]H2OpH6.8恩格列净结晶型0.101mg/mL0.108mg/mL恩格列净/富马酸1.85mg/mL1.78mg/mL恩格列净/柠檬酸12.2mg/mL11.8mg/mL恩格列净/L-焦谷氨酸2.45mg/mL2.36mg/mL(恩格列净共结晶体和恩格列净结晶型的溶解度)如上述表3所示确认到,相比于公知的恩格列净结晶型,本发明的恩格列净/富马酸共结晶体的水溶解度及小肠pH6.8下的溶解度增加约18倍,无定形形态的恩格列净/柠檬酸共结晶体的水溶解度及小肠pH6.8下的溶解度增加约120倍以上,恩格列净/L-焦谷氨酸共结晶体的水溶解度及小肠pH6.8下的溶解度增加约24倍。因此,确认到本发明的恩格列净共结晶体相比于公知的恩格列净结晶型,水溶解度可增加到约120倍。因此,本发明的恩格列净共结晶体是可克服成为恩格列净结晶型的问题的低水溶解度的新的结晶形态,这预测到可使恩格列净的药效极大化。[实验例4]恩格列净共结晶体和恩格列净结晶型的加速、苛刻稳定性比较评价所谓医药品的稳定性试验是指为了设置医药品等的储存方法及使用期限,设置适当规格之后,根据所定的试验法判断显著变化,来设置保质期的过程,故而确保药物的适当稳定性是在药物的产品化中尤为重要的因素之一。因此,为了确认本发明的恩格列净/富马酸共结晶体、无定形形态的恩格列净/柠檬酸共结晶体、恩格列净/L-焦谷氨酸共结晶体的产品化可能性,将国际公开专利公报WO2006/117359号中公知的恩格列净结晶型作为对照组,根据ICH指导原则,实施加速及苛刻稳定性试验,利用美国药典(USP)中记载的高效液相色谱(HPLC)分析法来分析之后,将其结果示于表4和5中。[表4]初期3天7天恩格列净/富马酸99.84%99.82%99.83%恩格列净/柠檬酸99.82%99.83%99.82%恩格列净/L-焦谷氨酸99.84%99.83%99.83%恩格列净结晶型99.8%99.32%99.02%(恩格列净共结晶体和恩格列净结晶型的加速稳定性结果,40℃±2℃,RH75%)[表5]初期3天7天恩格列净/富马酸99.84%99.81%99.82%恩格列净/柠檬酸99.82%99.82%99.82%恩格列净/L-焦谷氨酸99.84%99.84%99.83%恩格列净结晶型99.8%99.02%98.07%(恩格列净共结晶体和恩格列净的苛刻稳定性评价结果,60℃±2℃,RH75%)实施根据实施例制备的恩格列净共结晶体和恩格列净结晶型的加速、苛刻条件的稳定性试验。其结果如表4和5所示,确认到虽然恩格列净共结晶体不受纯度影响而保持稳定,但是恩格列净结晶型的纯度降低,稳定性差。因此,确认到恩格列净共结晶体的加速、苛刻条件下的稳定性比起恩格列净结晶型更加得到改善。[实验例5]恩格列净共结晶体和恩格列净结晶型的水溶解性比较试验为了增加体内吸收率,水溶解性应当要增加一些。因此,按1mg/mL的浓度,对本发明的恩格列净共结晶体和公知的恩格列净结晶型的水溶解性进行比较评价。其结果确认到,恩格列净结晶型未被溶解,以悬浮液状态存在,反之,本发明的恩格列净共结晶体均被溶解,水溶解性增加。因此,证明了本发明的共结晶体通过改善水溶解度来可克服恩格列净的难溶性的新的原料医药品价值[图15]。故而,本发明恩格列净共结晶体期待克服成为恩格列净的问题的低稳定性及水溶解度的新的原料医药品的可能性。以上,详细描述了本发明的特定部分,这些具体描述只不过是优选实例,而且本发明的范围并不限定于此,这对于本领域的普通技术人员来说是显而易见的。因此,本发明的实质性范围应当根据所附的权利要求书和其等同技术方案做出定义。当前第1页12
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。