一种阳离子缺陷型zif类多孔材料及其制备方法与应用
技术领域
1.本发明属于功能性吸附材料的技术领域,尤其涉及一种阳离子缺陷型zif类多孔材料及其制备方法与应用。
背景技术:
2.核能作为一种绿色、可靠的能源,随着世界范围内能源需求的增加,其重要性也日益突显。然而,放射性核废料释放到环境中的危害也是核能工业中药品管理要考虑的重要问题之一。在众多的放射性元素中,放射性碘具有独特的暴露问题。碘单质(i2)由于极易升华(45℃)而具有高流动性,它的同位素
129
i及
131
i也存在独特的暴露问题。
129
i是一种极其长寿的同位素,其半衰期为1.57
×
107年,在其衰变的过程中必须捕获与可靠的存储,而
131
i的半衰期仅为8.02天,但需即刻捕获,因其与人类的代谢过程有关。放射性碘可对人类代谢过程造成不利损害。这些放射性同位素吸入或通过食物链摄取后,往往会在甲状腺中富集,释放出破坏性的射线,导致肺癌,并提高癌症的病发率。
3.在过去的几十年里,人们研究了一些碘吸附剂,如二氧化硅,活性炭,沸石等。目前,工业上常用的方法是将有机胺(如三乙胺)浸渍在多孔材料(如活性炭)中,所制得的吸附剂对放射性碘具有较高的亲和力和净化效果,且捕获碘的过程相对简单。近年来,一些新型的多孔材料,如金属
‑
有机框架(metal
‑
organic framework,mof),共价
‑
有机框架(covalent
‑
organic framework,cof)已被证明具有较强的碘的吸附及存储功能。同时,这些新材料由于其孔道结构及大小可调,比表面积大,弱极性的材料空腔而备受关注。
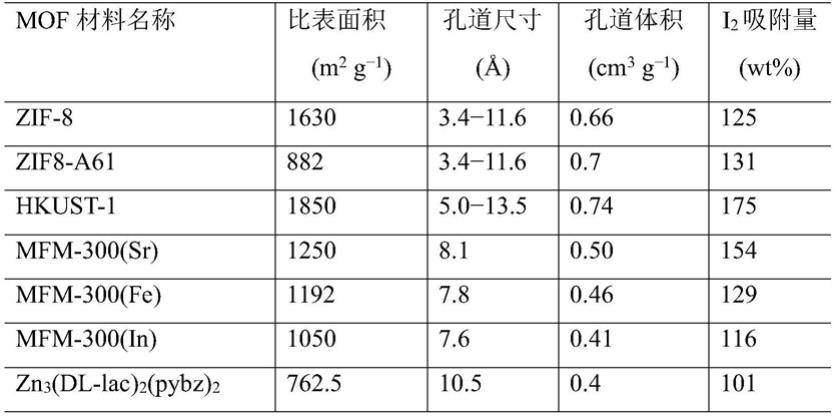
4.例如,hkust
‑
1(hong kong university ofscience and technology)具高的比表面积(1850m
2 g
‑1)及孔道尺寸在75
‑
80℃及常压下可以吸附175wt%的i2(表1)。然而,该材料由于孔道的开口尺寸远远高于i2(约),这给长期存储具有弱极性的i2带来困难。另外,mfm
‑
300(sr)/mfm
‑
300(fe)/mfm
‑
300(in)以及zn3(dl
‑
lac)2(pybz)2等也由于孔道的开口尺寸较大造成i2的长期存储困难(表1)。同时mfm
‑
300系列材料及zn3(dl
‑
lac)2(pybz)2等还由于需要使用价格高昂的四羧酸及吡啶羧酸配体,且mof材料制备条件苛刻等,这给这些材料走向实际应用造成困难。因此,选取廉价易得的合成原料,通过温和绿色的反应条件而得到结构稳定、i2存储量高、存储时间长的mof存储材料是此类材料走向实际应用所要解决的关键问题。
5.表1
[0006][0007]
在诸多mof材料中,具有沸石结构的zif
‑
8(zif=zeolitic imidazolate framework)由于具有合适的孔径、大的比表面积、高的化学稳定性和热稳定性,是一种理想的i2捕集剂。zif
‑
8由四面体配位zn
2
与2
‑
甲基咪唑(meim)配体连接而成的一个具有方钠石拓扑的三维网格结构(结果见附图1)。该结构中,β
‑
环的直径为通过直径为的六元环孔连接。该六元环孔的存在使得i2(约)可进行扩散并被有效封存。另外,结构中较小的四元环孔由于尺寸较小而不允许任何客体i2进入或逃离孔洞。
[0008]
例如,nenoff和navrotsky等人的研究发现,zif
‑
8对i2的最大吸附量高达125wt%,其中,25wt%i2与zif
‑
8表面结合,而剩余100wt%i2则被有效地封存zif
‑
8的孔洞中,并仅随着框架分解(约300℃)才被释放。baek等人通过后修饰的手段将zif
‑
8中meim替换为3
‑
胺基
‑
1,2,4
‑
三唑配体(atz),当atz的含量达到61%时,所得材料zif8
‑
a61对i2的吸附量可高达zif
‑
8的8.7倍。
[0009]
现有材料对于放射性i2的吸附及存储主要存在以下诸多方面的问题或不足:(1)存储量低(如活性炭);(2)材料孔道较大,不利于长期保存(如zn3(dl
‑
lac)2(pybz)2);(3)所用合成mof材料的有机配体成本较高、合成条件苛刻(如mfm
‑
300系列);(4)i2仅以单质(i2)形式存储,不利于长期保存(如zif
‑
8);(5)由于非极性i2与吸附剂的结合作用较弱,结合位点不稳定,可导致碘进一步释放,现有的捕集方法较难牢固地固定i2。
[0010]
目前报道的大多数是通过利用纳米金属有机框架(nanoscale metal
‑
organic framework;nmof)的孔洞来包埋抗癌药,抗癌药在运输的过程中存在泄露的风险,不仅导致抗癌药的毒副作用显现出来,而且还会大大降低抗癌的效果。此外,大多数nmof不易进入肿瘤细胞内部,需要用聚乙二醇、透明质酸或者叶酸等对其表面进行修饰,经过修饰的nmof就能较容易的穿过磷脂双分子层进入细胞,然而这就会增加合成难度和成本。例如在2017年,li课题组将抗癌药阿霉素和维拉帕米包裹在zif
‑
8中,为了能够靶向并进入癌细胞,于是在zif
‑
8的表面修饰了叶酸,zif
‑
8表面的叶酸与肿瘤细胞表面的叶酸受体进行特异性结合,从而使得纳米粒子能轻易进入肿瘤细胞。
[0011]
此外,由于单一的治疗模式杀死肿瘤细胞的效果有限,所以需要多种治疗模式相结合,例如光动力治疗和化疗相结合、放疗和化疗相结合等。例如在2019年,zhang课题组报道了将阿霉素(dox)和光敏剂二氢卟吩e6(ce6)包埋在zif
‑
8中,到达肿瘤细胞内,dox和ce6
同时释放,在630nm的光源照射下,ce6会产生活性氧实现光动力治疗,与此同时,dox发挥着化疗的作用(图2)。
[0012]
现有纳米粒子载体运输药物大多是通过简单的物理包埋或者吸附,在未到达肿瘤部位药物就会从载体上脱落,因此药物负载量就会降低。此外,大多数纳米粒子的生物兼容性差,不易穿过肿瘤细胞的细胞膜,导致肿瘤细胞内化率低。另外,绝大多数纳米粒子对肿瘤细胞或者细胞器没有靶向性,需要利用带有靶向功能基团的修饰,这就导致合成难度大。与此同时,由于单一的治疗模式对肿瘤细胞的致死率较低,所以需要多种治疗模式相结合,例如化疗和光动力治疗相结合,才能对肿瘤细胞具有较高的致死率,这导致合成的成本过高。
[0013]
针对现有技术中技术问题:包括肿瘤细胞靶向、肿瘤的毒性、sirna的携带、抗癌药的运输以及纳米粒子的合成成本等问题,急需一种新型技术方案解决该问题。
技术实现要素:
[0014]
为解决上述技术问题,本发明提供了一种阳离子缺陷型zif类多孔材料及其制备方法与应用。本发明在zif
‑
8的合成过程中加入含有“n”甲基化形成“n
‑
ch
3”基团的2
‑
甲基咪唑。该基团中仅sp2杂化的n被甲基化形成季铵阳离子,而sp3杂化的n仍然保留nh形式,通过碱性条件下与zn的配位形成
‑
1价离子以平衡化合价,整个配体呈端基配位(见图3)。因此可以通过构建基于2
‑
甲基咪唑的zif
‑
8中含有甲基化的咪唑阳离子缺陷形成富含i
‑
的阳离子型zif
‑
8,所得的化合物命名为zif
‑8
‑
i
‑
。
[0015]
一种阳离子缺陷型zif类多孔材料的制备方法,包括以下步骤:
[0016]
步骤(1):在有机溶剂中,将咪唑类化合物与碘甲烷混合反应得到咪唑基阳离子配体;
[0017]
步骤(2):将步骤(1)中所得咪唑基阳离子配体与咪唑类化合物溶解在水中,并加入金属盐混合均匀,静置反应,得到所述阳离子缺陷型zif类多孔材料。
[0018]
在本发明的一个实施例中,步骤(1)中,所述有机溶剂为四氢呋喃、二甲基甲酰胺、二氧六环、丙酮、二甲亚砜、甲醇、乙醇、乙腈、苯和甲苯中的一种或几种。
[0019]
在本发明的一个实施例中,步骤(1)和步骤(2)中,所述咪唑类化合物选自咪唑、苯并咪唑、2
‑
甲基咪唑、咪唑
‑2‑
甲醛、2
‑
氯
‑
咪唑和2
‑
苯基咪唑中的一种或几种。
[0020]
在本发明的一个实施例中,步骤(1)中,所述碘甲烷与咪唑类化合物摩尔比为1:1
‑
3:1。
[0021]
在本发明的一个实施例中,步骤(1)中,反应时间为8
‑
24小时,反应温度为55
‑
75℃。
[0022]
在本发明的一个实施例中,步骤(2)中,所述咪唑基阳离子配体与咪唑类化合物的摩尔比为1:30
‑
1:20。
[0023]
在本发明的一个实施例中,步骤(2)中,所述金属盐为醋酸锌、硝酸锌、乙酸钴和硝酸钴中的一种或几种。
[0024]
在本发明的一个实施例中,步骤(2)中,所述金属盐与咪唑类化合物的摩尔比为1:40
‑
1:70。
[0025]
如上述制备方法所得阳离子缺陷型zif类多孔材料。
[0026]
如上述阳离子缺陷型zif类多孔材料在碘吸附中的应用。
[0027]
本发明的上述技术方案相比现有技术具有以下优点:
[0028]
本发明所述的阳离子型材料zif
‑
8
‑
i
‑
的成功合成,将i
‑
作为材料不可分割的一部分固定于材料的孔洞中,可以继续与i2形成i3‑
,同时也可发挥zif
‑
8自身高效的i2吸附性能,最终达到物理及化学吸附协同吸附的效果。通过改进zif
‑
8形成阳离子型zif
‑8
‑
i
‑
,本发明成功地避免了i2的单一物理吸附同时通过将i2转化为i3‑
,大大提升了存储的稳定性。
[0029]
本发明制备得到材料原料廉价易得、合成绿色简单、i2的存储量大且保存时间长等。
[0030]
本发明合成的具有缺陷的阳离子型zif
‑
8(zif
‑8
‑
i
‑
)可以通过静电作用力与某些阴离子型抗癌药物相结合,这就使得药物在运输的过程中不易从载体上脱落下来,并且zif
‑8
‑
i
‑
在酸性的肿瘤环境中易分解,因此药物可以实现在肿瘤部位完全释放出来,从而实现高效的抗癌效果。带正电荷药物载体可以促进与带负电荷的细胞膜的相互作用,从而提高了癌细胞的内化速度和程度,此次合成的zif
‑8
‑
i
‑
是带有正电荷的纳米金属有机框架,能很好的与细胞膜相互作用,因此就能比较容易的进入肿瘤细胞,并且合成的步骤比较简单,成本低。
[0031]
除此之外,本发明还对zif
‑8
‑
i
‑
的细胞毒性进行研究,实验发现随着zif
‑8
‑
i
‑
浓度的不断增加,肿瘤细胞的活力呈阶梯式降低,由此看出zif
‑8
‑
i
‑
对肿瘤细胞存在明显的毒性,因此用该纳米粒子作为载体杀死肿瘤细胞的效率会增加。
附图说明
[0032]
为了使本发明的内容更容易被清楚地理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中
[0033]
图1是zif
‑
8的合成过程及结构示意图。
[0034]
图2是zif8
‑
a61的合成过程示意图。
[0035]
图3是本发明的zif
‑8
‑
i
‑
的合成示意图。
[0036]
图4是本发明咪唑基阳离子配体1h nmr。
[0037]
图5是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8三种材料的pxrd对比图。
[0038]
图6是本发明实施例1中所得zif
‑
8
‑
i
‑
(30:1)的eds谱图。
[0039]
图7是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8三种材料的1h nmr的谱图。
[0040]
图8是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8三种材料的扫描电镜图。
[0041]
图9是本发明实施例1中所得zif
‑8
‑
i
‑
(30:1)红外吸收谱图。
[0042]
图10是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8三种材料的热重曲线。
[0043]
图11是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8三种材料的n2的吸脱附曲线。
[0044]
图12是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8三种材料在正己烷中对碘的吸附曲线。
[0045]
图13是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8三种材料对碘蒸气的吸附曲线。
[0046]
图14是本发明zif
‑8
‑
i
‑
(20:1)、zif
‑8
‑
i
‑
(30:1)与zif
‑
8材料的细胞毒性实验结果。
具体实施方式
[0047]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。本发明中所述zif
‑
8是根据文献dalton trans.,2013,42,11128
–
11135制备所得。
[0048]
实施例1
[0049]
1)咪唑基阳离子配体的合成
[0050]
首先,将2
‑
甲基咪唑(411mg,5mmol)加入到反应瓶中,接着加入四氢呋喃(20ml)使2
‑
甲基咪唑完全溶解,最后加入碘甲烷(710mg,5mmol),并且升温至65℃反应过夜,反应结束后可以看到大量的淡黄色咪唑基阳离子配体从四氢呋喃析出,将产物离心分离,并用四氢呋喃洗涤多次除去多余的碘甲烷,并干燥得到第一步产物。1h nmr(400mhz,d2o)δ7.36
–
7.27(m,2h),3.79(d,j=0.9hz,3h),2.61(d,j=9.5hz,3h),结果见图4,由核磁数据看出,在位移值为3.79处有一个甲基的单峰,是2
‑
甲基咪唑上的“n”被甲基化形成“n
‑
ch
3”基团,由此证明了目标产物的合成。
[0051]
2)zif
‑8
‑
i
‑
的合成
[0052]
称取2
‑
甲基咪唑(1230mg,15mmol)与咪唑基阳离子型配体(112mg,0.5mmol),使它们完全溶解在5ml的去离子水中。另外称取醋酸锌(110mg,0.5mmol)并溶解在5ml的去离子水中,最后将两种溶液充分混合,静置5h,最终得到乳白色溶液,离心分离(10,000r/min)10min,并用去离子水洗涤三次,并冷冻干燥得到最终产物zif
‑8
‑
i
‑
(30:1)。
[0053]
实施例2
[0054]
1)咪唑基阳离子配体的合成
[0055]
首先,将2
‑
甲基咪唑(411mg,5mmol)加入到反应瓶中,接着加入四氢呋喃(20ml)使2
‑
甲基咪唑完全溶解,最后加入碘甲烷(710mg,5mmol),并且升温至65℃反应过夜,反应结束后可以看到大量的淡黄色咪唑基阳离子配体从四氢呋喃析出,将产物离心分离,并用四氢呋喃洗涤多次除去多余的碘甲烷,并干燥得到第一步产物。
[0056]
另称取2
‑
甲基咪唑(1230mg,15mmol)与咪唑基阳离子型配体(168mg,0.75mmol),使它们完全溶解在5ml的去离子水中。另外称取醋酸锌(83mg,0.38mmol)并溶解在5ml的去离子水中,最后将两种溶液充分混合,静置5h,最终得到乳白色溶液,离心分离(10,000r/min)10min,并用去离子水洗涤三次,并冷冻干燥得到最终产物zif
‑8
‑
i
‑
(20:1)。
[0057]
实施例3
[0058]
1)咪唑基阳离子配体的合成
[0059]
首先,将咪唑(341mg,5mmol)加入到反应瓶中,接着加入二甲基甲酰胺(20ml)使咪唑完全溶解,最后加入碘甲烷(710mg,5mmol),并且升温至55℃反应24h,反应结束后可以看到大量的淡黄色咪唑基阳离子配体从二甲基甲酰胺析出,将产物离心分离,并用二甲基甲酰胺洗涤多次除去多余的碘甲烷,并干燥得到第一步产物。
[0060]
2)zif
‑8
‑
i
‑
的合成
[0061]
称取咪唑(1022mg,15mmol)与咪唑基阳离子型配体(210mg,1mmol),使它们完全溶
解在5ml的去离子水中。另外称取硝酸锌(89mg,0.3mmol)并溶解在5ml的去离子水中,最后将两种溶液充分混合,静置5h,最终得到乳白色溶液,离心分离(10,000r/min)10min,并用去离子水洗涤三次,并冷冻干燥得到最终产物zif
‑8
‑
i
‑
(15:1)。
[0062]
实施例4
[0063]
1)咪唑基阳离子配体的合成
[0064]
首先,将咪唑
‑2‑
甲醛(480mg,5mmol)加入到反应瓶中,接着加入二甲亚砜(20ml)使咪唑
‑2‑
甲醛完全溶解,最后加入碘甲烷,(1420mg,10mmol),并且升温至65℃反应10h,反应结束后可以看到大量的淡黄色咪唑基阳离子配体从二甲亚砜析出,将产物离心分离,并用二甲亚砜洗涤多次除去多余的碘甲烷,并干燥得到第一步产物。
[0065]
2)zif
‑8
‑
i
‑
的合成
[0066]
称取咪唑
‑2‑
甲醛(1441mg,15mmol)与咪唑基阳离子型配体(90mg,0.38mmol),使它们完全溶解在5ml的去离子水中。另外称取乙酸锌(55mg,0.25mmol)并溶解在5ml的去离子水中,最后将两种溶液充分混合,静置5h,最终得到乳白色溶液,离心分离(10,000r/min)10min,并用去离子水洗涤三次,并冷冻干燥得到最终产物zif
‑8
‑
i
‑
(40:1)。
[0067]
实施例5
[0068]
1)咪唑基阳离子配体的合成
[0069]
首先,将2
‑
氯
‑
咪唑(513mg,5mmol)加入到反应瓶中,接着加入甲苯(20ml)使2
‑
氯
‑
咪唑完全溶解,最后加入碘甲烷(2130mg,15mmol),并且升温至75℃反应24h,反应结束后可以看到大量的淡黄色咪唑基阳离子配体从甲苯析出,将产物离心分离,并用甲苯洗涤多次除去多余的碘甲烷,并干燥得到第一步产物。
[0070]
2)zif
‑8
‑
i
‑
的合成
[0071]
称取2
‑
氯
‑
咪唑(1538mg,15mmol)与咪唑基阳离子型配体(73mg,0.3mmol),使它们完全溶解在5ml的去离子水中。另外称取硝酸锌(60mg,0.2mmol)并溶解在5ml的去离子水中,最后将两种溶液充分混合,静置8h,最终得到乳白色溶液,离心分离(10,000r/min)10min,并用去离子水洗涤三次,并冷冻干燥得到最终产物zif
‑8
‑
i
‑
(50:1)。
[0072]
实施例6
[0073]
1)咪唑基阳离子配体的合成
[0074]
首先,将苯并咪唑(591mg,5mmol)加入到反应瓶中,接着加入甲醇(20ml)使苯并咪唑完全溶解,最后加入碘甲烷(2130mg,15mmol),并且升温至75℃反应24h,反应结束后可以看到大量的淡黄色咪唑基阳离子配体从甲苯析出,将产物离心分离,并用甲苯洗涤多次除去多余的碘甲烷,并干燥得到第一步产物。
[0075]
2)zif
‑8
‑
i
‑
的合成
[0076]
称取苯并咪唑(1772mg,15mmol)与咪唑基阳离子型配体(78mg,0.3mmol),使它们完全溶解在5ml的去离子水中。另外称取硝酸锌(60mg,0.2mmol)并溶解在5ml的去离子水中,最后将两种溶液充分混合,静置8h,最终得到乳白色溶液,离心分离(10,000r/min)10min,并用去离子水洗涤三次,并冷冻干燥得到最终产物zif
‑8
‑
i
‑
(50:1)。
[0077]
对比例1
[0078]
根据文献dalton trans.,2013,42,1112811135制备得到zif
‑
8:称取2
‑
甲基咪唑(1230mg,15mmol),使其完全溶解在5ml的去离子水中。另外称取醋酸锌(110mg,0.5mmol)并
溶解在5ml的去离子水中,最后将两种溶液充分混合,静置5h,最终得到乳白色溶液,离心分离(10,000r/min)10min,并用去离子水洗涤三次,并冷冻干燥得到最终产物zif
‑
8。
[0079]
材料表征
[0080]
将实施例1制备得到的zif
‑8
‑
i
‑
(30:1)和实施例2制备得到的zif
‑8
‑
i
‑
(20:1)以及对比例1中制备得到的zif
‑
8进行性能表征,其中包括:x
‑
射线粉末衍射(pxrd)(见图5)、eds能谱(见图6)、核磁共振(见图7)、扫描电子显微镜(sem)(见图8)和傅里叶变换红外(ft
‑
ir)(见图9)等充分表征,确定了具有缺陷的阳离子型zif
‑8
‑
i
‑
的合成。
[0081]
由图5可知,zif
‑8
‑
i
‑
(30:1)、zif
‑8
‑
i
‑
(20:1)与zif
‑
8的pxrd的结果基本一致,这就表明掺杂进入的咪唑基阳离子配体不会降低材料的结晶性。
[0082]
由图6可知,由于产物存在“i
‑”,从图中可以看出产物中存在碘元素的,测试结果与预测一致,因此表明zif
‑8
‑
i
‑
(30:1)的成功合成。
[0083]
由图7可知,将zif
‑8
‑
i
‑
用60μl的氘代盐酸溶解,再加入0.6ml的氘代二甲亚砜测试核磁共振。可以看出,掺杂不同量的咪唑基阳离子配体,在位移值为3.68处存在甲基的单峰,这是2
‑
甲基咪唑上的“n”被甲基化形成“n
‑
ch
3”基团,由此看出咪唑基阳离子有机配体成功的掺杂进入zif
‑
8的骨架中,形成产物zif
‑8
‑
i
‑
,并且随着掺杂量的增大,核磁的峰随之增高。
[0084]
由图8可知,两种zif
‑8
‑
i
‑
(30:1)、zif
‑8
‑
i
‑
(20:1)的形貌和尺寸相比于纯zif
‑
8基本没有发生改变,因此zif
‑8
‑
i
‑
基本能维持zif
‑
8的形貌。
[0085]
图9为zif
‑8
‑
i
‑
(30:1)的红外光谱及数据:2926(w),1570(w),1458(w),1422(w),1310(m),1178(m),1146(s),993(m),953(w),847(w),758(s),692(m)cm
‑1。
[0086]
图10为热重曲线,由图可知,zif
‑
8在500℃开始失重,而zif
‑8
‑
i
‑
在150℃开始失重,失重到80%时停止失重,推测是由于咪唑基阳离子的配体掺杂到骨架中导致zif
‑8
‑
i
‑
结构不稳定,直到500℃再次开始失重,这与zif
‑
8保持一致。
[0087]
图11为三种材料对n2的吸脱附曲线,由图可知,与原始的zif
‑
8相比,zif
‑8
‑
i
‑
样品的brunauer
–
emmett
–
teller(bet)比表面积和孔体积随着咪唑基阳离子型配体的掺杂量的增加而逐渐减小。虽然咪唑基阳离子配体的掺杂导致了这些性质的下降,但zif
‑8
‑
i
‑
样品的空穴和大表面积足以吸附i2分子(见表2),
[0088]
表2 zif
‑8
‑
i
‑
(30:1)、zif
‑8
‑
i
‑
(20:1)、zif
‑
8的比表面积和孔径
[0089]
absorbentss
bet
(m2·
g
‑1)v
pore
(cm3·
g
‑1)zif
‑
816372.79zif
‑8
‑
i
‑
(30:1)13882.13zif
‑8
‑
i
‑
(20:1)12701.83
[0090]
性能测试例
[0091]
测试例1
[0092]
1)zif
‑
8、zif
‑8
‑
i
‑
(30:1),以及zif
‑8
‑
i
‑
(20:1)在正己烷中对i2的吸附实验。
[0093]
分别称取zif
‑
8、zif
‑8
‑
i
‑
(30:1),以及zif
‑8
‑
i
‑
(20:1)三种材料各10mg,再配制3000mg/l的碘的正己烷溶液,分别取10ml溶液加入到3个样品瓶中,将三种材料加入到溶液中,静置。分别间隔0h、2h、6h、9h、12h、24h和48h取出0.5ml的上清液,并用正己烷稀释到5ml,用紫外分光光度计测得溶液在400nm和600nm之间的紫外吸收绘制曲线并计算碘的吸
附量。实验结果见图13。
[0094]
图12中,图12
‑
a、12
‑
b和12
‑
c分别是zif
‑
8、zif
‑8
‑
i
‑
(30:1)和zif
‑8
‑
i
‑
(20:1)在碘的正己烷中,不同时间上清液的紫外吸收图,通过观察发现,相比于zif
‑
8,zif
‑8
‑
i
‑
上清液的紫外吸光度随着时间的增加从而迅速下降,这就意味着zif
‑8
‑
i
‑
材料对正己烷中的碘有着高效的吸附作用。通过计算得出12
‑
d图,从数据上看,zif
‑8
‑
i
‑
(20:1)材料在正己烷中对碘的吸附高达2200mg/g,这比zif
‑
8在正己烷中对碘的吸附高了大约1000mg/g,因此zif
‑8
‑
i
‑
显示出高效的碘吸附功能。
[0095]
测试例2
[0096]
1)zif
‑
8、zif
‑8
‑
i
‑
(30:1),以及zif
‑8
‑
i
‑
(20:1)在对i2蒸气的吸附实验。
[0097]
分别称取分别称取zif
‑
8、zif
‑8
‑
i
‑
(30:1),以及zif
‑8
‑
i
‑
(20:1)三种材料10mg放入到样品中,再称取大量的碘单质加入到样品瓶中,将他们同时放入到广口瓶中盖上盖子,将广口瓶放入到75℃恒温烘箱。分别间隔0h、1h、3h、6h、9h、12h、24h、36h和48h称量装有材料的样品瓶,最终计算出碘蒸气吸附量。实验结果见图14。
[0098]
由图13可以看出,zif
‑8
‑
i
‑
相对于zif
‑
8对碘蒸气的吸附略微增加,并没有与在正己烷中对碘的吸附增加效果明显,这可能是溶液和蒸汽两种吸附过程的机理不同导致的。
[0099]
测试例3
[0100]
为了研究zif
‑8
‑
i
‑
的抗癌功效,进行了细胞毒性试验,首先选取了黑色素瘤细胞(b16f10)和脑胶质瘤细胞(u87)两个细胞系作为研究对象。分别称取5.18mg纯的zif
‑
8和5.21mg的zif
‑8
‑
i
‑
(30:1),将两种材料分别完全分散在5ml的去离子水中,取上述制备溶液4ml加入相应的pbs干粉(12.4mg
·
ml
‑1),超声溶解铺板:每孔150μl,4复孔。最浓一组直接用pbs缓冲液后用原液逐级稀释2倍(无血清dmem),zif
‑
8和zif
‑8
‑
i
‑
浓度依次为62.5μg/ml、125μg/ml、250μg/ml、500μg/ml,孵育24h,加入mtt 100μl,4h后翻板,加入二甲亚砜100μl。摇床上震荡3min,用酶标仪测试570nm处的吸光度,得到相应的存活率。
[0101]
实验结果:由图14可知,在b16f10细胞系中随着纯zif
‑
8浓度增加,肿瘤细胞的活力几乎没有降低,因此纯zif
‑
8对b16f10细胞不存在明显毒性,但是随着zif
‑8
‑
i
‑
浓度的不断增加,肿瘤细胞的活力呈阶梯式降低。值得注意的是当zif
‑8
‑
i
‑
的浓度为500μg/ml时,肿瘤细胞的活力降低到了40%以下,因此zif
‑8
‑
i
‑
对b16f10细胞存在明显的毒性。在u87细胞系中随着纯的zif
‑
8浓度增加,肿瘤细胞的活力略微降低,说明zif
‑
8对u87细胞系存在微弱毒性。但是随着zif
‑8
‑
i
‑
浓度的不断增加,肿瘤细胞的活力迅速降低。值得注意的是当浓度为500μg/ml时,可使u87肿瘤细胞全死(当细胞活力为20%左右可视为全死),因此zif
‑8
‑
i
‑
对u87细胞存在较大毒性,以上实验结果表明zif
‑8
‑
i
‑
存在明显的抗癌功效,是一种具有较大潜力的抗癌材料。
[0102]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。