1.本发明属于有机化合物制备技术领域,具体涉及一种肟菌酯特征杂质的制备方法。
背景技术:
2.肟菌酯(trifloxystrobin)是由先正达公司研制、德国拜耳公司开发的第二代甲氧丙烯酸酯类杀菌剂,具有广谱、耐雨水冲刷性能好、持效期长和对作物安全等优点,对作物安全,其结构式如下:
[0003][0004]
由于肟菌酯在土壤、水中可快速降解,因此对环境的安全性高;而且,肟菌酯是具有化学动力学特性的杀菌剂,它能被植物蜡质层强烈吸附,对植物表面提供优异的保护活性。肟菌酯的适宜作物包括小麦、大麦、黑麦、黑小麦、葡萄、苹果、花生、香蕉和蔬菜等,应用范围较广。肟菌酯主要用于茎叶处理,保护活性强,且活性不受环境影响,其应用最佳期为孢子萌发和发病初期阶段,但对黑星病的各个时期均有活性。
[0005]
肟菌酯自问世以来就得到了广泛的关注和应用,其制备工艺的研发也成为农药化学领域的研究热点。目前比较常见的肟菌酯制备路线如下所示:
[0006][0007]
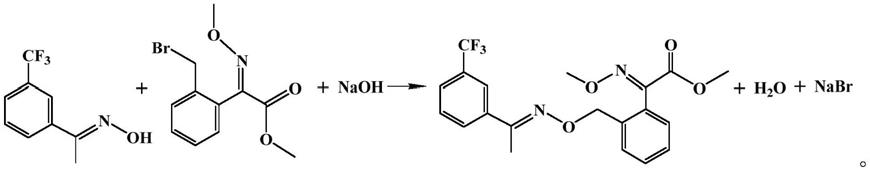
在上述工艺路线中,反应在碱作用下进行缩合,由于该反应中碱与(e)-2-(2-溴甲基苯基)-2-甲氧亚胺基乙酸甲酯均过量,而碱性环境下肟菌酯不可避免地会发生皂化反应生成中间体盐(2e)-2-甲氧基亚氨基-2-[2-[[1-[3-(三氟甲基)苯基]亚乙基氨基]氧甲基]苯基]乙酸钠,即肟菌酸钠。在后续的酯化反应中,肟菌酸钠与(e)-2-(2-溴甲基苯基)-2-甲氧亚胺基乙酸甲酯(溴代肟醚)发生反应,生成肟菌酯特征杂质(e)-2-((e)-2-甲氧基-1-(甲氧亚氨基)-2-乙氧基)苯基-2-(甲氧亚氨基)-2-(2-(((e)-1-(3-(三氟甲基)苯基)亚乙基氨基)甲氧基)苯基)乙酸甲酯,该特征杂质的结构如下所示:
[0008]
[0009]
肟菌酯特征杂质在制备肟菌酯原药过程中作为副产物出现,由于肟菌酯特征杂质的性质与肟菌酯原药十分相似,难以去除,严重影响了肟菌酯原药的纯度和品质。为了提高肟菌酯原药的产品品质,该特征杂质的分析与控制就十分重要,但是,控制肟菌酯原药的质量就必须要求对杂质进行检测,即需要这种特征杂质作为对照品。pasumpon kamaraj等采用一锅法合成了上述肟菌酯特征杂质,但是该合成方法中使用了大量碱,且碱与溴代肟醚同时加入会使部分溴代肟醚发生分解,从而致使目标产物的收率偏低(pasumpon kamaraj等,letters in organic chemistry,2015,12,306-310)。
[0010]
因此,开发一种简单实用、绿色环保的肟菌酯特征杂质的合成方法,以得到高收率和高纯度的肟菌酯特征杂质,对于进一步控制肟菌酯的产品质量、扩大市场推广具有重要的意义。
技术实现要素:
[0011]
针对现有技术的不足,本发明的目的在于提供一种肟菌酯特征杂质的制备方法,所述制备方法以肟菌酯为起始原料,通过皂化反应和缩合反应得到目标产物,所述制备方法的条件温和、工艺简单,可以得到高收率和高纯度的肟菌酯特征杂质,能够作为对照品应用于肟菌酯分析方法的研发中。
[0012]
为达此目的,本发明采用以下技术方案:
[0013]
本发明提供一种肟菌酯特征杂质的制备方法,所述制备方法包括如下步骤:
[0014]
(1)肟菌酯与碱进行皂化反应,得到肟菌酸盐,反应式如下:
[0015][0016]
(2)将步骤(1)得到的肟菌酸盐与溴代肟醚进行缩合反应,生成的粗产物进行后处理,得到所述肟菌酯特征杂质,反应式如下:
[0017][0018]
其中,m选自钾或钠。
[0019]
本发明所述制备方法的目标产物为肟菌酯特征杂质,其化学名称为(e)-2-((e)-2-甲氧基-1-(甲氧亚氨基)-2-乙氧基)苯基-2-(甲氧亚氨基)-2-(2-(((e)-1-(3-(三氟甲基)苯基)亚乙基氨基)甲氧基)苯基)乙酸甲酯。所述制备方法以肟菌酯为起始原料,首先将肟菌酯通过皂化反应生成中间体盐肟菌酸盐,该步骤中皂化反应的程度高,反应后几乎所有的肟菌酯被转化为肟菌酸盐;而后加入溴代肟醚,肟菌酸盐与溴代肟醚发生缩合生成目标产物,缩合反应中肟菌酸盐的转化程度高,无其他副产物发生,反应效果好;将缩合反应得到的粗产物进行后处理,最终目标产物的产率可达到87%以上,甚至达到96.4%,高效液相色谱法(hplc)检测含量达到98.5%以上。通过本发明提供的制备方法得到的肟菌酯特征
杂质产率高、纯度高,可作为对照品,有利于肟菌酯分析方法的开发,更有利于肟菌酯的产品质量控制,为产品质量控制提供了新的思路。
[0020]
本发明中,步骤(1)所述肟菌酯与碱的摩尔比为1:(1.0~1.2),例如1:1.01、1:1.03、1:1.05、1:1.07、1:1.09、1:1.1、1:1.11、1:1.13、1:1.15、1:1.17或1:1.19等。
[0021]
优选地,所述m为钠,即步骤(1)所述碱优选为氢氧化钠。
[0022]
本发明中,步骤(1)所述皂化反应的温度为30~90℃,例如33℃、35℃、38℃、40℃、42℃、45℃、48℃、50℃、52℃、55℃、58℃、60℃、62℃、65℃、68℃、70℃、72℃、75℃、78℃、80℃、82℃、85℃、87℃或89℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0023]
优选地,步骤(1)所述皂化反应的时间为1~5h,例如1.1h、1.3h、1.5h、1.7h、1.9h、2h、2.2h、2.5h、2.8h、3h、3.2h、3.5h、3.8h、4h、4.2h、4.5h、4.7h或4.9h,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0024]
本发明中,步骤(1)所述皂化反应在溶剂中进行。
[0025]
优选地,所述溶剂与肟菌酯的质量比为(3~5):1,例如3.1:1、3.3:1、3.5:1、3.7:1、3.9:1、4:1、4.1:1、4.3:1、4.5:1、4.7:1或4.9:1等。
[0026]
优选地,所述溶剂为n,n-二甲基甲酰胺(dmf)。
[0027]
优选地,步骤(1)所述皂化反应在搅拌条件下进行。
[0028]
本发明中,步骤(2)所述肟菌酸盐与溴代肟醚的摩尔比为1:(1.0~1.5),例如1:1.03、1:1.05、1:1.08、1:1.1、1:1.12、1:1.15、1:1.18、1:1.2、1:1.22、1:1.25、1:1.28、1:1.3、1:1.33、1:1.35、1:1.38、1:1.4、1:1.42、1:1.45、1:1.47或1:1.49等。
[0029]
本发明中,步骤(2)所述缩合反应的温度为30~90℃,例如33℃、35℃、38℃、40℃、42℃、45℃、48℃、50℃、52℃、55℃、58℃、60℃、62℃、65℃、68℃、70℃、72℃、75℃、78℃、80℃、82℃、85℃、87℃或89℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0030]
优选地,步骤(2)所述缩合反应的时间为6~24h,例如6.5h、7h、7.5h、8h、8.5h、9h、9.5h、10h、10.5h、11h、11.5h、12h、12.5h、13h、13.5h、14h、14.5h、15h、15.5h、16h、16.5h、17h、17.5h、18h、18.5h、19h、19.5h、20h、20.5h、21h、21.5h、22h、22.5h、23h或23.5h,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0031]
本发明中,步骤(2)所述缩合反应在溶剂中进行。
[0032]
优选地,所述溶剂为n,n-二甲基甲酰胺。
[0033]
本发明中,步骤(2)所述后处理的方法为:将粗产物脱溶,得到的有机相经萃取、酸化、重结晶,得到所述肟菌酯特征杂质。
[0034]
优选地,所述萃取的萃取剂为二氯甲烷和/或二氯乙烷,进一步优选为二氯乙烷。
[0035]
优选地,所述萃取剂与有机相的质量比为(3~6):1,例如3.1:1、3.3:1、3.5:1、3.7:1、3.9:1、4:1、4.1:1、4.3:1、4.5:1、4.7:1、4.9:1、5:1、5.2:1、5.4:1、5.6:1、5.8:1或5.9:1等。
[0036]
优选地,所述酸化的酸性试剂为盐酸和/或硫酸,进一步优选为盐酸。
[0037]
优选地,所述重结晶的溶剂为甲醇或甲醇水溶液,进一步优选为甲醇。
[0038]
优选地,所述甲醇水溶液中甲醇的质量百分含量≥80%,例如81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、92%或94%等。
[0039]
优选地,所述重结晶后进行固液分离,收集固相,液相浓缩后再次结晶;合并固相,得到所述肟菌酯特征杂质。
[0040]
优选地,所述后处理的方法具体为:将粗产物脱溶,得到的有机相i与萃取剂和水混合后,向其中加入酸性试剂、直至体系中无白色固体析出;分层后将有机相ii脱溶,与重结晶的溶剂混合溶解后进行重结晶,固液分离,将固相干燥,液相浓缩后再次结晶;合并固相,得到所述肟菌酯特征杂质。
[0041]
优选地,所述酸性试剂的加入方式为滴加。
[0042]
优选地,所述酸性试剂为过量添加,即至体系中无白色固体析出后,再滴加少量酸性试剂。
[0043]
优选地,所述重结晶的温度为0~10℃,例如0.5℃、1℃、1.5℃、2℃、2.5℃、3℃、3.5℃、4℃、4.5℃、5℃、5.5℃、6℃、6.5℃、7℃、7.5℃、8℃、8.5℃、9℃或9.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0044]
优选地,所述制备方法具体包括如下步骤:
[0045]
(1)肟菌酯与碱在溶剂中30~90℃皂化反应1~5h,得到肟菌酸盐,所述肟菌酯与碱的摩尔比为1:(1.0~1.2);
[0046]
(2)将步骤(1)得到的肟菌酸盐与溴代肟醚以摩尔比1:(1.0~1.5)混合,在溶剂中30~90℃进行缩合反应6~24h,生成粗产物;将所述粗产物脱溶,得到的有机相经萃取、酸化、重结晶,得到所述肟菌酯特征杂质。
[0047]
相对于现有技术,本发明具有以下有益效果:
[0048]
本发明提供的制备方法以肟菌酯为起始原料合成肟菌酯特征杂质,所述制备方法首先将肟菌酯皂化生成肟菌酸盐,得到的肟菌酸盐进一步与溴代肟醚发生缩合反应,生成目标产物。所述制备方法通过皂化和缩合两步反应快速合成肟菌酯特征杂质,工艺步骤简单,反应条件温和,易于操作,原料转化率高,无副反应发生;经过后处理得到的最终产物纯度大于98.5%,产率可达到87%以上。通过本发明提供的制备方法得到的肟菌酯特征杂质具有高纯度和高产率特点,可作为对照品,有利于肟菌酯分析方法的开发,更有利于肟菌酯的质量控制,为产品质量控制提供了新的思路。
具体实施方式
[0049]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0050]
本发明以下实施例中的原料肟菌酯可通过现有技术制备得到;产物纯度通过高效液相色谱(hplc,lc-20at,日本岛津)外标法测得,收率为摩尔收率,计算公式为:y=(m1×
m2×
ω2)/(m1×
0.98
×
m2)
×
100%,其中,y为收率,m1为肟菌酯的相对分子质量,m1为肟菌酯的投入量,m2为肟菌酯特征杂质的相对分子质量,m2为肟菌酯特征杂质的质量,ω2为肟菌酯特征杂质的hplc检测含量(即纯度)。
[0051]
实施例1
[0052]
本实施例提供一种肟菌酯特征杂质(e)-2-((e)-2-甲氧基-1-(甲氧亚氨基)-2-乙氧基)苯基-2-(甲氧亚氨基)-2-(2-(((e)-1-(3-(三氟甲基)苯基)亚乙基氨基)甲氧基)苯基)乙酸甲酯的制备方法,具体步骤如下:
[0053]
(1)肟菌酸钠的合成
[0054][0055]
向反应釜中加入620.1g dmf和206.9g肟菌酯(外标含量为98%),开动搅拌,随后升温到30℃,缓慢加入21.8g氢氧化钠(96%,0.525mol),反应5h后hplc检测反应完毕后(归一原料<0.5%,主峰>98%),得反应液i。
[0056]
(2)肟菌酯特征杂质的合成
[0057][0058]
向步骤(1)得到的反应液i中加入分批次加入共174.8g溴代肟醚(90%,0.55mol),于30℃下进行缩合反应,取样检测,至肟菌酸钠消耗完毕视为反应结束,共反应24h,脱溶回收dmf,得釜内有机相i。
[0059]
(3)肟菌酯特征杂质的后处理
[0060]
向步骤(2)得到的有机相i中加入1000.0g二氯甲烷和200.3g水,室温下缓慢滴加60%硫酸至无白色固体析出后,再加少量硫酸。分层后保留有机相ii,并将有机相ii脱溶,然后加1200.0g质量百分含量为80%的甲醇水溶液,加热溶解后冷却至0℃结晶,过滤,收集固相并烘干,得目标产物216.7g,hplc外标法检测含量为99.4%;滤液浓缩后继续结晶,得含量为98.6%的目标产物64.4g;合并目标产物,反应总收率为93.7%。
[0061]
目标产物的检测:通过液相色谱-质谱联用法(lc/ms)测得[m 1]
(%):600(100%);
[0062]1h-nmr(cdcl3,400mhz):δ2.12(s,3h,ch3),3.82(s,3h,ch3),4.02(s,6h,ch3),5.12(s,4h,ch2),7.15-7.28(m,2h,arh),7.33-7.41(m,5h,arh),7.45(m,1h,arh),7.57(m,2h,arh),7.76(m,1h,arh),7.83(m,1h,arh)。
[0063]
实施例2
[0064]
本实施例提供一种肟菌酯特征杂质(e)-2-((e)-2-甲氧基-1-(甲氧亚氨基)-2-乙氧基)苯基-2-(甲氧亚氨基)-2-(2-(((e)-1-(3-(三氟甲基)苯基)亚乙基氨基)甲氧基)苯基)乙酸甲酯的制备方法,具体步骤如下:
[0065]
(1)肟菌酸钠的合成
[0066]
[0067]
向反应釜中加入784.3g dmf和207.1g肟菌酯(外标含量为98%),开动搅拌,随后升温到60℃,缓慢加入20.8g氢氧化钠(96%,0.5mol),反应2h后hplc检测反应完毕后(归一原料<0.5%,主峰>98%),得反应液i。
[0068]
(2)肟菌酯特征杂质的合成
[0069][0070]
向步骤(1)得到的反应液i中加入分批次加入共190.9g溴代肟醚(90%,0.6mol),于60℃下进行缩合反应,取样检测,至肟菌酸钠消耗完毕视为反应结束,共反应10h,脱溶回收dmf,得釜内有机相i。
[0071]
(3)肟菌酯特征杂质的后处理
[0072]
向步骤(2)得到的有机相i中加入1000.6g二氯乙烷和200.7g水,室温下缓慢滴加盐酸至无白色固体析出后,再加少量盐酸。分层后保留有机相ii,并将有机相ii脱溶,然后加1200.0g甲醇(≥99.5%),加热溶解后冷却至0℃结晶,过滤,收集固相并烘干,得目标产物231.2g,hplc外标法检测含量为99.6%;滤液浓缩后继续结晶,得含量为98.8%的目标产物57.8g;合并目标产物,反应总收率为96.4%。
[0073]
目标产物的检测:lc/ms测得[m 1]
(%):600(100%);
[0074]1h-nmr(cdcl3,400mhz):δ2.12(s,3h,ch3),3.82(s,3h,ch3),4.02(s,6h,ch3),5.12(s,4h,ch2),7.15-7.28(m,2h,arh),7.33-7.41(m,5h,arh),7.45(m,1h,arh),7.57(m,2h,arh),7.76(m,1h,arh),7.83(m,1h,arh)。
[0075]
实施例3
[0076]
本实施例提供一种肟菌酯特征杂质(e)-2-((e)-2-甲氧基-1-(甲氧亚氨基)-2-乙氧基)苯基-2-(甲氧亚氨基)-2-(2-(((e)-1-(3-(三氟甲基)苯基)亚乙基氨基)甲氧基)苯基)乙酸甲酯的制备方法,具体步骤如下:
[0077]
(1)肟菌酸钠的合成
[0078][0079]
向反应釜中加入1000.4g dmf和206.5g肟菌酯(外标含量为98%),开动搅拌,随后升温到90℃,缓慢加入25.1g氢氧化钠(96%,0.6mol),反应1h后hplc检测反应完毕后(归一原料<0.5%,主峰>98%),得反应液i。
[0080]
(2)肟菌酯特征杂质的合成
[0081]
[0082]
向步骤(1)得到的反应液i中加入分批次加入共238.8g溴代肟醚(90%,0.75mol),于90℃下进行缩合反应,取样检测,至肟菌酸钠消耗完毕视为反应结束,共反应6h,脱溶回收dmf,得釜内有机相i。
[0083]
(3)肟菌酯特征杂质的后处理
[0084]
向步骤(2)得到的有机相i中加入1000.4g二氯乙烷和201.7g水,室温下缓慢滴加盐酸至无白色固体析出后,再加少量盐酸。分层后保留有机相ii,并将有机相ii脱溶,然后加1187.4g质量百分含量为90%的甲醇水溶液,加热溶解后冷却至0℃结晶,过滤,收取固相并烘干,得目标产物191.1g,hplc外标法检测含量为99.6%;滤液浓缩后继续结晶,得含量为99.0%的目标产物69.8g;合并目标产物,反应总收率为87.3%。
[0085]
目标产物的检测:lc/ms测得[m 1]
(%):600(100%);
[0086]1h-nmr(cdcl3,400mhz):δ2.12(s,3h,ch3),3.82(s,3h,ch3),4.02(s,6h,ch3),5.12(s,4h,ch2),7.15-7.28(m,2h,arh),7.33-7.41(m,5h,arh),7.45(m,1h,arh),7.57(m,2h,arh),7.76(m,1h,arh),7.83(m,1h,arh)。
[0087]
申请人声明,本发明通过上述实施例来说明本发明的一种肟菌酯特征杂质的制备方法,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。