血清中维生素k1的高效液相色谱串联质谱检测方法
技术领域
1.本发明涉及生物医药检测技术领域,特别是涉及一种血清中维生素k1的高效液相色谱串联质谱检测方法。
背景技术:
2.维生素k1(vk1),又称甲萘醌,是一种脂溶性维生素,其化学性质较稳定,能耐酸、耐热,但对光敏感,也易被碱和紫外线分解。vk1是肝脏合成凝血因子过程中所必需物质,当其缺乏时会影响凝血因子ⅱ、
ⅶ
、
ⅸ
、
ⅹ
的合成,造成凝血酶原复合物生成减少并出现自发性出血,具有潜在致命风险。vk1状态可以用于评估新生儿出血性疾病,乳糜泻,克罗恩病,溃疡性结肠炎和慢性胰腺炎(囊性纤维化)等吸收不良疾病,亦有报道表明其与骨质密度或阿尔茨海默病亦存在关联。因此,建立有效的vk1检测方法十分重要。
3.由于人体内存在多种脂溶性维生素,包括维生素a、维生素d、维生素e、维生素k等,且血液样本本身具有复杂多样性,采用色谱质谱分析的方法进行脂溶性维生素分析测定时,干扰因素特别多,基质干扰特别严重。对于如何能够充分提取、纯化目标化合物,并尽可能少的引入不必要的基质干扰,以满足项目检测高灵敏度、高特异性的要求是一个非常重要和复杂的关键过程。同时体内不同脂溶性维生素含量不同(维生素a、维生素e浓度相对较高)、极性差异大,若要准确的测定vk1,还要尽可能的实现色谱分离,以免其他维生素对vk1的检测造成干扰。特别是vk1在血液中的含量低至pmol/l,并在体内常与蛋白紧密结合,因此,对方法的检测灵敏度要求极高。此外血清样本十分复杂,除了具有大量的蛋白质、盐以及磷脂外,人体产生的代谢产物也可能会对目标物的检测造成干扰。另外vk1在有光的条件下不稳定的性质也需要予以考虑。
4.目前对于vk1常用的检测方法有高效液相-紫外检测法、高效液相-荧光检测法、气相色谱串联质谱法等。但仍存在前处理过程复杂、基质干扰严重,灵敏度低等问题。因此,为了更好的指导维生素k1的补充治疗,亟待开发可以准确定性和定量、除杂效果好、灵敏度更高的vk1检测方法。
技术实现要素:
5.基于此,本发明的目的是提供一种步骤简单,且特异性强、灵敏度高、抗干扰能力好的血清中维生素k1的高效液相色谱串联质谱检测方法。
6.具体技术方案如下:
7.一种血清中维生素k1的高效液相色谱串联质谱检测方法,包括以下步骤:
8.样品前处理:在待测血清样本中加入沉淀剂进行沉淀;再加入萃取剂进行萃取,离心;将所得上清液干燥后用复溶液进行复溶,离心;再取所得上清液作为待测样品溶液;其中,所述沉淀剂包括2,6-二叔丁基-4-甲基苯酚、雌三醇、内标d
7-vk1和乙醇;所述萃取剂包括2,6-二叔丁基-4-甲基苯酚、雌三醇和正己烷;所述复溶液包括2,6-二叔丁基-4-甲基苯酚、雌三醇和甲醇;
9.液相色谱分离:将所述待测样品溶液使用高效液相色谱进行色谱分离;
10.质谱检测:将高效液相色谱分离后的样品进行质谱检测。
11.在其中一些实施例中,所述液相色谱分离的色谱条件包括:
12.流动相:有机相为2.5
±
0.3mm甲酸铵甲醇溶液,水相为含0.1
±
0.02v/v%甲酸的2.5
±
0.3mm甲酸铵水溶液;采用梯度洗脱,流速为0.5
±
0.1ml/min。
13.在其中一些实施例中,所述梯度洗脱包括:
14.0~0.5min:有机相70%~75%,水相25%~30%;
15.0.5~4min:有机相70%~75%
→
84~87%,水相25%~30%
→
13%~16%;
16.4~4.01min:有机相84~87%
→
88~90%,水相13%~16%
→
10~12%;
17.4.01~9min:有机相88~90%
→
98~100%,水相10~12%
→
0~2%;
18.9~10.5min:有机相98~100%,水相0~2%;
19.10.5~10.6min:有机相98~100%
→
70%~75%,水相0~2%
→
25%~30%;
20.10.6~12min:有机相70%~75%,水相25%~30%。
21.在其中一些实施例中,所述液相色谱分离的色谱条件包括:
22.色谱柱:asentis express c18;
23.洗针液:体积分数40~60%的甲醇水溶液;
24.柱温:40~50℃;
25.进样量:20
±
5μl。
26.在其中一些实施例中,所述色谱柱的长度为2~10cm,内径为2.1~4.6mm,粒径为2.7μm。
27.在其中一些实施例中,所述质谱检测采用ab sciex api 5500三重四极杆质谱仪。
28.在其中一些实施例中,所述质谱检测的条件包括:离子源:大气压化学电离源,正离子扫描模式,多反应监测模式;气帘气为40.0
±
5psi;碰撞气为5.0
±
0.5psi;雾化电流为3.0
±
0.5μa;雾化气为30.0
±
5psi;离子源温度为450.0
±
20℃。
29.在其中一些实施例中,维生素k1的定性离子对为:451.4/199.2;维生素k1的定量离子对为:451.4/187.2;d7-vk1内标离子对为458.5/194.1。
30.在其中一些实施例中,所述沉淀剂中2,6-二叔丁基-4-甲基苯酚的浓度为0.2~1.0mg/ml,雌三醇的浓度为0.8~1.6μg/ml,所述内标d
7-vk1的浓度为0.8~1.6ng/ml;
31.所述萃取剂中2,6-二叔丁基-4-甲基苯酚的浓度为0.2~1.0mg/ml,雌三醇的浓度为0.8~1.6μg/ml;
32.所述复溶液中2,6-二叔丁基-4-甲基苯酚的浓度为0.2~1.0mg/ml,雌三醇的浓度为0.8~1.6μg/ml。
33.在其中一些实施例中,所述待测血清样本、沉淀剂和萃取剂的体积比为1:3~5:7~9。
34.在其中一些实施例中,萃取后离心所得上清液与所述复溶液的体积比为6~8:1。
35.在其中一些实施例中,所述沉淀的过程包括漩涡震荡5~10min和室温孵育10~15min;所述萃取的过程包括漩涡震荡10~15min;所述复溶的过程包括漩涡震荡5~10min。
36.在其中一些实施例中,配制标准曲线工作溶液时加入空白基质,所述的空白基质为2~6wt%牛血清蛋白。
37.与现有技术相比,本发明具有以下有益效果:
38.本发明方法采用高效液相色谱-质谱联用,结合特定的样品前处理步骤、液相色谱条件和质谱检测条件,实现了一种特异性强、灵敏度高、抗基质干扰能力好、具有很好的重现性的血清中维生素k1检测方法。尤其本方法的灵敏度相比于已报道的方法更高(定量限更低),能覆盖更广泛的人群样本检测,特别是针对凝血功能异常的患者,有利于开展临床上对血清中vk1含量进行监测,可以用于诊断vk1相关的疾病,为临床治疗提供服务。
39.本发明的前处理步骤可以去除大部分极性较强的杂质,净化效果好,提取回收率高。并且,将内标溶解在沉淀剂中作为内标工作液一次性加入,将加内标和蛋白沉淀操作步骤合并,节省操作步骤,且不影响蛋白沉淀效果和提取回收率。
40.本发明的前处理试剂中(沉淀剂、萃取剂和复溶液),所添加的2,6-二叔丁基-4-甲基苯酚和雌三醇复配能在目标物萃取及转移过程中起到保护作用,有利于提高检测灵敏度和目标物的响应信号,增强处理后样本的稳定性,避免样本不稳定带来的操作问题,减少vk1以及内标在萃取以及转移过程中的损失,减少样本量;复溶液中使用甲醇作为溶剂,对目标物溶解性好,离子化效率高,vk1绝对响应值更高,且不存在溶剂效应。
41.对于前处理过程中未能去除的杂质和性质相似的代谢物,本发明进一步对液相色谱流动相种类、梯度洗脱、空白基质等进行优化,降低基质干扰,消除基质效应,不仅实现了血清样本中vk1的有效分离和定量,还能实现对多种脂溶性维生素的色谱分离,可扩展应用到准确定量分析。
42.在上述基础上,本发明进一步对质谱条件进行了优化,克服了vk1极性较弱、检测灵敏度低的缺陷,实现低样本用量(100μl)就可以达到高灵敏度(定量限低至0.038ng/ml,即84.4pmol/l)的效果。
附图说明
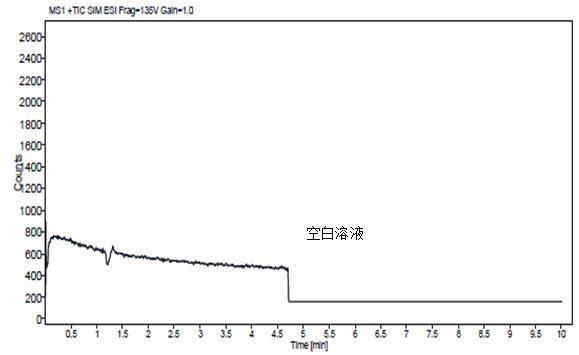
43.图1为实施例1中1例血清样本中vk1的总离子流色谱图;
44.图2为vk1的线性回归曲线;
45.图3为前处理过程中未加入保护剂处理的vk1检测色谱图;
46.图4为前处理过程中加入保护剂处理的vk1检测色谱图;
47.图5为组a的色谱图;
48.图6为组b的色谱图;
49.图7为组c的色谱图;
50.图8为样本中vk1及其他脂溶性维生素色谱图;
51.图9为混合血清紫外照射前的色谱图;
52.图10为混合血清紫外照射60h后的色谱图;
53.图11为bsa空白基质的色谱图。
具体实施方式
54.本发明下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例中所用到的各种常用化学试剂,均为市售产品。
55.除非另有定义,本发明所使用的所有的技术和科学术语与属于本发明的技术领域
的技术人员通常理解的含义相同。本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不用于限制本发明。
56.本发明的术语“包括”和“具有”以及它们任何变形,意图在于覆盖不排他的包含。例如包含了一系列步骤的过程、方法、装置、产品或设备没有限定于已列出的步骤或模块,而是可选地还包括没有列出的步骤,或可选地还包括对于这些过程、方法、产品或设备固有的其它步骤。
57.在本发明中提及的“多个”是指两个或两个以上。“和/或”,描述关联对象的关联关系,表示可以存在三种关系,例如,a和/或b,可以表示:单独存在a,同时存在a和b,单独存在b这三种情况。字符“/”一般表示前后关联对象是一种“或”的关系。
58.以下结合具体实施例对本发明作进一步详细的说明。
59.实施例1
60.(1)溶液配制:
61.雌三醇储备液的配制:称取一定量雌三醇固体,用无水乙醇溶解,制备得到浓度为1.0mg/ml的雌三醇储备液。
62.内标中间液的配制:用无水乙醇稀释维生素k1内标(d
7-vk1)储备液,得到浓度为10.0μg/ml的内标中间液。
63.沉淀剂的配制:分别吸取40μl-80μl的内标中间液及400μl-800μl的雌三醇储备液于500ml容量瓶中,再加入0.1g-0.5g bht,用无水乙醇定容得到沉淀剂。
64.萃取剂的配制:吸取400μl-800μl的雌三醇储备液至500ml容量瓶,再加入0.1g-0.5g bht,用正己烷定容得到萃取剂。
65.复溶液的配制:加入0.1g-0.5g bht和400μl-800μl的雌三醇储备液至500ml容量瓶,用纯甲醇定容得到复溶液。
66.4%bsa溶液配制:称取一定量牛血清蛋白(bsa)固体,用去离子水溶解,得到4%(w/w)bsa溶液。
67.vk1标准品中间液ⅰ:用无水乙醇将1.0mg/ml的vk1标准品储备液稀释成浓度为10.0μg/ml的中间液ⅰ。
68.vk1标准品中间液ⅱ:以4%bsa作为空白曲线基质将中间液ⅰ稀释成50.0ng/ml的中间液ⅱ。
69.(2)样品前处理方法:
70.1)移取100μl血清样本,加入400μl沉淀剂,漩涡震荡5~10min;
71.2)室温孵育10~15min;加入800μl萃取剂,漩涡震荡10~15min;
72.3)然后在转速为10000~12000rpm下离心5~10min;
73.4)吸取上层萃取液700μl转至2.0ml离心管中,室温氮气吹干;
74.5)向吹干离心管中加入100μl复溶液,漩涡震荡5~10min,在转速为10000-12000rpm、25℃离心3min;
75.6)取上清液转移至棕色进样瓶,作为待测样品,待上机检测。
76.(3)待测样品中vk1的检测
77.将待测样品使用液相色谱-质谱仪进行检测:
78.1、液相色谱条件:色谱柱采用asentis express c18(5cm*2.1mm,2.7μm);流动相a
(有机相)为含有2.5mm甲酸铵甲醇溶液,流动相b(水相)为含有0.1%甲酸~2.5mm甲酸铵水溶液;洗针液为体积分数50%的甲醇水溶液,采用梯度洗脱模式,液相梯度如表1所示。柱温为45℃,进样量为20μl。
79.表1 液相洗脱梯度
80.步骤分析时间(min)流速(ml/min)流动相a%流动相b%100.5732720.50.57327340.5861444.010.58911590.51000610.50.51000710.60.573278120.57327
81.2、质谱检测采用ab sciex api 5500三重四极杆质谱仪,大气压化学电离源(apci),正离子扫描模式,多反应监测模式(mrm)。质谱条件包括:气帘气为40.0psi;碰撞气为5.0psi;雾化电流为3.0μa;雾化气为30.0psi;离子源温度为450.0℃。mrm扫描模式具体质谱参数如表2所示。
82.表2 质谱参数
[0083][0084]
(4)血清中vk1的定性、定量检测
[0085]
以样品中vk1两个离子对与标准品的化合物保留时间(rt为8.72min)进行比对作为定性依据。用内标标准曲线法定量,vk1的峰面积与d
7-vk1峰面积的比值为纵坐标,vk1的浓度与d
7-vk1的浓度比值为横坐标,加权方式为权重1/x,绘制标准曲线,即可对实际样本进行定量计算。
[0086]
(5)待测样品检测
[0087]
采用上述方法,对1例血清样本中的vk1进行了含量测定,检测总离子流色谱图如图1所示。从图中可看出,vk1的色谱峰(rt为8.72min)峰型对称,响应合适,分离效果很好。
[0088]
(6)方法学验证
[0089]
1、线性关系和定量限:
[0090]
以4wt%bsa溶液为空白基质,对vk1标准品中间液ⅱ(50ng/ml)进行稀释,配制得到浓度分别为0.038ng/ml、0.076ng/ml、0.15ng/ml、0.30ng/ml、0.60ng/ml、1.21ng/ml、2.42ng/ml、4.83ng/ml、9.66ng/ml的一系列标准工作曲线溶液。按样品前处理方法每天将上述每个浓度的曲线样本平行处理2个,检测3天,计算各浓度样本的平均值、rsd和回收率,
结果见表3。图2为vk1的线性回归曲线(回归方程为:y=0.14729x 0.00143,r=0.99905)。结果表明vk1在线性范围0.038ng/ml~9.66ng/ml(84.36pmol/l~21445pmol/l)内各浓度点rsd<20%,回收率均在85%~115%之内,且具有良好的线性关系。vk1定量限为0.038ng/ml(84.36pmol/l),检出限为0.0100ng/ml(22.2pmol/l)。
[0091]
表3 线性数据
[0092][0093][0094]
2、精密度试验:
[0095]
收集混合病人血清样本或病人基质加标得到低、中、高不同浓度水平(浓度分别在0.30ng/ml、1.5ng/ml、6.5ng/ml附近)的样本,每个浓度同一天各平行处理12份,进行批内精密度实验,如表4结果表明三个不同浓度水平vk1的精密度rsd都在2.6%~3.3%之间,表明该方法的批内精密度良好。
[0096]
取上述混合病人血清样本或加标得到的低、中、高不同浓度水平(浓度分别在0.30ng/ml、1.5ng/ml、6.5ng/ml附近)的样本各20份,每个浓度每天测定两组,连续10天进行相同的前处理和检测,如表4结果表明三个不同浓度的血清样本中vk1的精密度rsd都在4.3%~6.2%之间,表明该方法的批间精密度良好。
[0097]
表4 批内、批间精密度实验结果
[0098]
[0099]
3、加标回收率:
[0100]
收集病人血清样本混合成低、中、高不同浓度(浓度分别在0.30ng/ml、0.90ng/ml、2.10ng/ml附近)的混合血清基质,分别加不同浓度标准品溶液进行加标回收率实验;加标前后的样品,按照本实施例中方法平行处理3次,计算加标样品结果回收率。结果如表5所示,表明此方法在高中低三个不同浓度水平范围的回收率在93.1%~108.2%之间,满足回收率要求,方法准确。
[0101]
表5 加标回收率结果
[0102][0103]
实施例2 样品前处理过程的优化
[0104]
vk1在光照条件下不稳定,在紫外光照射以及碱性条件下易被分解。同时由于vk1脂溶性较强,易被吸附在前处理过程中使用的耗材(离心管、进样瓶等)上。因此,减少前处理过程中vk1在的损失,对于获得准确定量结果以及提升灵敏度都非常关键。在本发明中,通过向前处理过程中的试剂添加保护剂(抗氧化剂bht),减少对光不稳定带来的降解。同时为避免vk1被塑料离心管或者玻璃瓶吸附,还添加类固醇激素(雌三醇),通过竞争吸附保护待测目标物。
[0105]
对比同一个样本前处理过程中,在沉淀剂、萃取剂及复溶液中是否添加保护剂(bht和雌三醇)对vk1的影响,平行处理三次结果均表明前处理过程中添加保护剂(bht以及雌三醇)检测得到的vk1响应信号更高(如图3~4所示),说明加入保护剂能够一定程度保护vk1,有利于提高vk1的检测灵敏度和稳定性。
[0106]
综上述结果,最终选择了在前处理过程中,所有使用的沉淀剂、萃取剂、复溶液中都加入bht和雌三醇保护目标物vk1。
[0107]
实施例3 样本处理后稳定性测试
[0108]
因vk1对光敏感不稳定,在处理过程中和处理后的稳定性都特别重要。收集病人样本混合成高低不同浓度(浓度在6.9ng/ml、0.8ng/ml附近)的混合血清,按照优化后的方法前处理过程中加入保护剂处理,分别将高、低混合血清样本各平行处理10份,将处理后的高、低浓度待测样分别混合在一起,然后用棕色玻璃进样瓶各分装成8份,在进样盘(15℃)条件下保存,分别于0、2、4、8、12、24、36、48h各测定3次,验证样本处理后的保存稳定性;以0h检测结果为参考靶值,其它不同存放时间点的检测结果与0h结果相比。如表6结果表明,回收率均在85%~115%之间,且3次平行测定的cv均在10%以内,说明样品在处理过程中加入保护剂后,在进样盘条件下可稳定放置至少48h。
[0109]
表6 处理后样品放置于进样盘(15℃)的稳定性试验数据
[0110][0111][0112]
实施例4 不同复溶液种类对vk1检测的影响
[0113]
由于在色谱质谱方法中,一般采用优化确定的色谱条件初始流动相作为复溶液,将前处理后的样本进行复溶后上机检测,主要是为了避免出现溶剂效应。但由于vk1极性较弱,且在水相中难溶,同时为了最大程度的保证其离子化效率,本实施例对复溶液的种类进行了研究,比较了两种不同的复溶液体系:纯甲醇溶液(复溶液1)和73%有机相-27%水相
(复溶液2,实施例1中流动相初始比例)。
[0114]
收集病人样本混合成不同浓度水平(浓度分别在0.35ng/ml、3.5ng/ml附近)的血清样本,分别按实施例1中前处理方法,平行处理三批次后进行测定(结果如表7所示)。
[0115]
表7 不同复溶液的比较
[0116][0117]
结果表明采用复溶液2(流动相初始比例溶液)作为复溶液,vk1及其内标d
7-vk1的峰面积均较低,其可能是目标物vk1及其内标未能完全溶解导致离子化效率降低。因此,本发明优选采用纯甲醇作为复溶液体系,vk1的峰面积更高,且不存在溶剂效应。
[0118]
实施例5 流动相条件筛选
[0119]
由于血液样本基质复杂,经过前处理纯化处理后,仍然会有一些杂质会干扰目标物vk1的测定,有些理化性质相似的化合物还会共流出影响vk1离子化效率。而有效的分离杂质和目标物有利于改善基质效应。因此,本发明设计了三组不同的流动相,考察流动相对目标物或者杂质的保留行为,对比不同的流动相组成对目标物vk1的色谱分离效果。
[0120]
组a:有机相为2.5mm甲酸铵甲醇溶液,水相为含0.1v/v%甲酸的2.5mm甲酸铵水溶液;
[0121]
组b:有机相为纯甲醇溶液,水相为纯水溶液;
[0122]
组c:有机相为纯甲醇溶液,水相为含0.1%甲酸的2.5mm甲酸铵水溶液。
[0123]
组a、b、c的色谱结果分别如图5~7所示。所选流动相基本都能将目标物实现色谱分离。但是,组a的流动相(有机相为2.5mm甲酸铵甲醇溶液,水相为含0.1%甲酸的2.5mm甲酸铵水溶液)和梯度洗脱程序,不仅能有效将杂质和目标物进行完全分离,且目标物出峰附近的色谱图中背景噪音较低。同时,该流动相和梯度程序条件下,本方法还能实现多种脂溶性维生素(维生素a、25-羟基维生素d2、25-羟基维生素d3、维生素e、维生素k1)的色谱分离(如图8所示),抗干扰性强,特异性高,可扩展为多种脂溶性维生素的定量方法。
[0124]
实施例6 空白基质选择和评估
[0125]
对于色谱质谱定量方法的建立以及基质效应的评估,配置标准曲线的空白基质选取十分关键和重要。一方面能够尽量模拟实际样品基质条件,另一方面所选基质不能存在明显的基质效应,不影响准确定量。本发明拟建立人体血清样本中vk1的检测方法,最佳的曲线基质应为人体空白血清基质。但vk1在人体血液中本身存在,即使根据vk1在紫外光照条件下不稳定、易被分解的原理,经过试验,将混合病人血清需放置在紫外灯下常温照射至少60h后,vk1本底才能去除干净(如图9、图10所示),预处理耗时较长。但是,本发明通过选择
牛血清白蛋白作为替代基质,经测定发现其vk1本底含量可忽略(如图11所示)。因此,基于bsa在节省预处理时间上的优势,我们采取bsa基质开展后续实验。
[0126]
基质效应评估:收集六个不同来源的病人样本(a1-a6),样本本底浓度范围在0.5~1.5ng/ml;同时使用4wt%bsa将实施例1中vk1标准品中间液ⅱ稀释配制成浓度分别为0.3ng/ml(l)、3.0ng/ml(h)的vk1标准溶液;分别将标准溶液l、h和六个不同来源的病人基质样本按1:1比例进行混合,得到混合样本al和ah;将标准溶液、病人样本和混合样本各平行处理三次,前处理结束后,将得到的待测样品转移至液相进样瓶中,用高效液相色谱三重四级杆串联质谱仪(lc-ms/ms)进行检测,通过vk1以及同位素内标d
7-vk1的峰面积,计算相应的比值(vk1/d
7-vk1峰面积之比)。通过混合样品(al、ah)的vk1/d
7-vk1比值与理论值(六个不同来源的病人样本a1-a6与低、高浓度的标准工作溶液响应的均值)的差异均小于20%,说明基质效应可忽略,结果如表8,表9所示。
[0127]
表8 基质效应结果(样本混合前)
[0128][0129]
表9 基质效应结果(样本混合后)
[0130]
[0131][0132]
综上所述,本发明方法采用高效液相色谱-质谱联用,结合特定的样品前处理步骤、液相色谱条件和质谱检测条件,实现了一种特异性强、灵敏度高、抗基质干扰能力好、具有很好的重现性的血清中维生素k1检测方法。尤其本方法的灵敏度相比于已报道的方法更高(定量限更低),能覆盖更广泛的人群样本检测,特别是针对凝血功能异常的患者,有利于开展临床上对血清中vk1含量进行监测,可以用于诊断vk1相关的疾病,为临床治疗提供服务。
[0133]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0134]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。