1.本发明属于药物化学技术领域,涉及一种医药中间体的制备,特别涉及一种吉非替尼中间体4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐的的制备方法。
背景技术:
2.吉非替尼(gefitinib)是阿斯利康公司研制的一种口服表皮生长因子受体酪氨酸激酶(egfr-tk)抑制剂。2002年在日本上市,用于治疗晚期非小细胞肺癌,2003年5月5日被fda批准作为局部晚期或转移性非小细胞肺的三线治疗药物。2005年以商品名易瑞沙在中国上市,用于治疗既往接受过化疗的局部晚期或转移性非小细胞癌。
3.吉非替尼有多条合成路线,主要是原研公司开发的三条路线(专利wo9633980)以及在此基础上进行的工艺优化和再开发。具体路线如下:路线一:该路线以6 ,7-二甲氧基喹唑啉-4(3h)酮为原料,用甲磺酸和l-蛋氨酸选择性脱甲基得到6-羟基-7-甲氧基-3 ,4-二氢喹唑啉-4-酮,然后在酚羟基上乙酰化后得关键中间体3 ,4-二氢-7-甲氧基-4-氧代喹唑啉-6-醇乙酸酯,再经氯代、胺化、水解、醚化得到目标产物吉非替尼。
4.路线二:
该路线以价廉易得的异香草醛为原料,经醚化、硝化、还原、水解及环化反应得到取代喹唑啉酮后,再与氯化亚砜氯代得到4-氯喹唑啉,然后以3-氯-4-氟苯胺芳胺亲核取代得到吉非替尼。
5.路线三:该路线同样以价廉易得的异香草醛为原料,经醚化、硝化、还原及dimroth重排反应环化反应得到吉非替尼。
6.上述方法各有优缺点,但是路线一中没有硝化反应等危险工艺,因此,路线一是制备吉非替尼安全且广泛的方法。其中,4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐是该路线关键中间体。我们对专利仔细研究分析,并结合实验情况,发现合成该中间体存在放大操作不便,杂质偏高等问题。蒸馏二氯亚砜,蒸干后物料容易结成块状,块状物里会包裹二氯亚砜等残留试剂,影响下一步反应,而且块状物很难从反应釜中取出,无法确定收率,同样对下一步投料产生影响。第二步为胺化反应,常用的溶剂是异丙醇,胺化反应的机理是3-氯-4-氟苯胺作为亲核试剂进攻6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐,从而发生
取代反应,然而溶剂异丙醇同样也有一定的亲核性,参与竞争反应,只是异丙醇的亲核性弱于3-氯-4-氟苯胺,因此,需要抑制异丙醇参与反应,减少副反应。这些问题会影响产品纯度和放大生产,因此,对该方法进行合理的改进尤为必要。
技术实现要素:
7.针对现有技术的缺陷,本发明的目的在于提供一种新的吉非替尼中间体4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐的制备方法,该方法条件温和,便于操作,适合车间放大生产。
8.本发明是通过以下技术方案实现的:吉非替尼中间体4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐的制备方法,包括如下步骤:步骤一、将6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮﹑二氯亚砜和n,n-二甲基甲酰胺置于反应釜中,加热反应,反应完毕,减压蒸馏至不再有液滴流出,加入有机溶剂,在一定温度下搅拌打浆,抽滤,得到6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐;步骤二、将6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐溶于异丙醇中,滴加3-氯-4-氟苯胺的异丙醇溶液,滴完后搅拌反应,反应结束后,抽滤得到吉非替尼中间体4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐。
9.上述过程的反应方程式为:本发明的进一步改进方案为:步骤一所述加热反应的温度为80~90℃,所述加热反应的时间为4~8h。
10.步骤一所述有机溶剂为结构中带有苯环的有机溶剂。
11.所述有机溶剂为甲苯﹑乙苯﹑甲氧基苯﹑氯苯﹑溴苯﹑二甲苯﹑三甲苯中的一种或两种以上混合。
12.步骤一中所述6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮与所述有机溶剂的质量比为1:3~15;所述6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮与所述二氯亚砜的质量比为1:5~10;所述6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮与所述n,n-二甲基甲酰胺的摩尔比为1:0.3~0.34。
13.步骤一所述打浆温度为20~60℃,所述打浆时间为40min~80min。
14.步骤二所述滴加温度为-10~50℃,所述反应温度为-10~20℃,所述反应时间为1~10h。
15.步骤二所述6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐与所述3-氯-4-氟苯胺的摩尔比为1:1.1~1.3。
16.本发明的有益效果为:(1)蒸馏二氯亚砜时会产生物料结块,蒸馏不彻底导致不好放料等情况,加入有机溶剂
打浆,可以分散物料,便于放料抽滤,同时可以去除包裹在块状物里的二氯亚砜等残留试剂,减少对下一步反应的影响。条件温和,便于操作,适合车间放大生产。
17.(2)第二步胺化反应,通过温度控制,可以抑制参与异丙醇反应,减少副反应,提高产品纯度。
附图说明
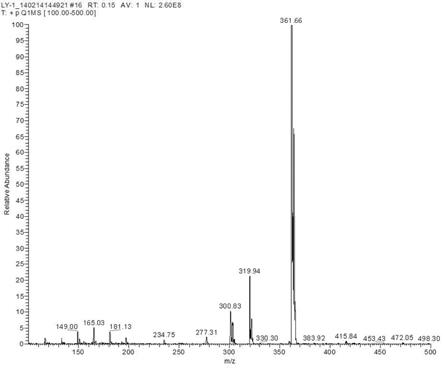
18.图1为实施例1制得的4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐的质谱图;图2为实施例1制得的4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐的核磁共振氢谱图。
具体实施方式
19.实施例1(1)将6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮(250g, 1.07mol)﹑二氯亚砜(1.75kg)和n,n-二甲基甲酰胺(25g, 0.34mol)置于3l反应瓶中,搅拌加热至回流,保温反应6h,反应完毕,降温,当内温降至60℃时,减压蒸馏, 至不再有液滴流出,加入甲苯(1.5kg),60℃时搅拌打浆1h,降温至20℃,抽滤,减压干燥得到280克 6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐,收率90.7%。
20.(2)将6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐(280g, 0.97mol)溶于异丙醇(1.5kg)中,降温至-5℃,滴加3-氯-4-氟苯胺(155g,1.07mol)的异丙醇溶液,-10~20℃反应5h,抽滤,减压干燥得到340克4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐,收率88.3%。
21.实施例2(1)将6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮(250g, 1.07mol)﹑二氯亚砜(1.75kg)和n,n-二甲基甲酰胺(25g, 0.34mol)置于3l反应瓶中,搅拌加热至回流,保温反应6h,反应完毕,降温,当内温降至60℃时,减压蒸馏, 至不再有液滴流出,加入乙苯(1.75kg), 50℃时搅拌打浆1h,降温至20℃,抽滤,减压干燥得到265克 6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐,收率85.8%。
22.(2)将6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐(265g, 0.86mol)溶于异丙醇(1.5kg)中,降温至-3℃,滴加3-氯-4-氟苯胺(150g,1.03mol)的异丙醇溶液,-10~20℃反应4h,抽滤,减压干燥得到300千克4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐,收率82.2%。
23.实施例3(1)将6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮(250g, 1.07mol)﹑二氯亚砜(1.75kg)和n,n-二甲基甲酰胺(25g, 0.34mol)置于3l反应瓶中,搅拌加热至回流,保温反应6h,反应完毕,降温,当内温降至60℃时,减压蒸馏, 至不再有液滴流出,加入甲氧基苯(1.5kg),40℃时搅拌打浆1h,降温至20℃,抽滤,减压干燥得到260克 6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐,收率84.3%。
24.(2)将6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐(260g, 0.90mol)溶于异丙醇
(1.5kg)中,降温至0℃,滴加3-氯-4-氟苯胺(170g,1.17mol)的异丙醇溶液,-10~20℃反应3h,抽滤,减压干燥得到290千克4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐。
25.实施例4(1)将6-乙酰氧基-7-甲氧基-3h喹唑啉-4-酮(5kg, 21.3mol)﹑二氯亚砜(35kg)和n,n-二甲基甲酰胺(0.5kg, 6.84mol)置于50l反应釜中,搅拌加热至回流,保温反应6h,反应完毕,降温,当内温降至60℃时,减压蒸馏, 至不再有液滴流出,加入甲苯(30kg),40℃时搅拌打浆1h,降温至20℃,抽滤,减压干燥得到5.7千克 6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐,收率92.4%。
26.(2)将6-乙酰氧基-4-氯-7-甲氧基喹唑啉盐酸盐(5.7kg, 19.7mol)溶于异丙醇(34.2kg)中,降温至2℃,滴加3-氯-4-氟苯胺(3.44kg,23.64mol)的异丙醇溶液,-10~20℃反应4h,抽滤,减压干燥得到6.8千克4-(3-氯-4-氟苯胺)-6-乙酰氧基-7-甲氧基喹唑啉盐酸盐,收率86.6%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
