1.本发明涉及胶黏剂技术领域,进一步地说,是涉及一种高强度光可逆胶粘 剂、制备方法及应用。
背景技术:
2.胶粘剂是一种能够将两块基材牢固粘接在一起的材料,广泛应用于军用与 民用的各个领域,是汽车,生物医学,建筑和航空航天业必不可少的材料。为 节约成本与减少环境污染,需要可逆胶粘剂设计与应用。
3.当前,可逆胶粘剂主要是基于热可逆机理,而光诱导方式一直因其非接触 式引发,无定向引发和快速简便的操作而受到关注。偶氮苯的光致异构化是一 种制备光诱导可逆胶粘剂的方式。含有连续偶氮苯序列的基质在可见光照射下 以固体的形式将基质结合在一起,在紫外光照射下液化以实现脱粘。而由于链 段运动能力的限制,仅适用于热塑性胶粘剂,且易发生蠕变变形。
4.tristan harper等人研究了一种含悬挂蒽基团的光诱导固化丙烯酸酯湿表面 胶粘剂,但在报道中没有包含对于胶粘剂可逆性能的研究。(t.harper,r.slegeris, i.pramudya,h.chung,single
‑
phase photo
‑
cross
‑
linkable bioinspired adhesive forprecise control of adhesion strength,acs appl.mater.interfaces.2017,9, 1830
–
1839.)其他研究者们还分别探索了蒽的衍生物在可逆热固性胶粘剂中的应 用。然而,这些粘合剂的应用除了在固化粘接过程中利用光诱导以外,在拆卸 过程中仍然依赖于加热辅助。
5.设计一种通过光引发来实现粘接与脱粘过程是很有意义的,这可以最大限 度地发挥光引发过程的优势。研究者们已经对粘接与拆卸过程中利用光诱导的 可逆热固性胶粘剂进行了研究。但现有的研究成果中仍然存在一个问题:当反应 产物的玻璃化转变温度明显高于环境温度时,聚合物链段在反应过程中被冻结, 导致反应效率低,甚至无法进行反应。具备较低玻璃化转变温度的体系虽然可 以保证固化反应的完成,但固化产物在室温下处于高弹态。上述两种情况均对 光诱导可逆胶粘剂的粘接强度产生不利影响。
技术实现要素:
6.为解决现有技术中出现的问题,本发明提供了一种高强度光可逆胶粘剂、 制备方法及应用。本发明的胶粘剂含有蒽基基团和聚己内酯结晶链段,是一种 具有优异粘接性能的光诱导可逆胶粘剂。本发明首先将含蒽基的化合物原料环 氧化,制备出含蒽的环氧化产物,再与端巯基醇化合物及促进剂混合,在室温 下进行开环,制备得到含有蒽基侧链的双端羟基化合物。采用双端异氰酸酯基 化合物和双端羟基的聚己内酯,加入有机锡催化剂,进行扩链反应形成较大分 子量的扩链双端异氰酸酯基化合物。最后,将扩链双端异氰酸酯基化合物与含 有蒽基侧链的双端羟基化合物和不含有蒽基侧链的双端羟基化合物反应,加入 有机锡催化剂催化反应,得到含有蒽侧基和聚己内酯结晶链段的线型聚氨酯结 构的
化合物,即所述的高强度光可逆胶粘剂。本发明的胶粘剂在350~405nm的 紫外光照射下进行[4 4]环加成反应,固化;在小于300nm的紫外光照射下解交 联。
[0007]
本发明的目的之一是提供一种高强度光可逆胶粘剂。
[0008]
所述胶粘剂包含以下结构:
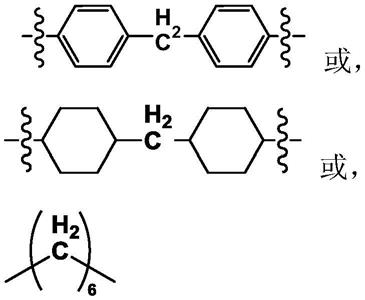
[0009][0010]
0<q<8;0<p<7;0<n<8;0<m<6;且p,q,n,m为整数;
[0011]
其中,
[0012]
a为
[0013][0014]
b为:
[0015][0016]
a为19~40之间的整数,包括端点19,40;
[0017]
d为:
[0018][0019]
b为4~6之间的整数,包括端点4,6;
[0020]
r为
[0021][0022]
本发明的胶粘剂在不同波长的紫外光照射下,能够发生如下反应:
[0023]
在350~405nm的紫外光照射下固化;优选在紫外光照射之前加热使胶粘剂 处于熔融状态;照射时间可以根据实际情况而定,一般为5
‑
10分钟;
[0024]
在小于300nm的紫外光照射下解交联;照射时间根据实际情况而定,一般 不少于2小时;
[0025][0026]
本发明的目的之二是提供一种高强度光可逆胶粘剂的制备方法。
[0027]
所述方法包括:
[0028]
步骤(1)、将蒽基化合物溶于溶剂a中,然后加入环氧化合物、碱和相转 移催化剂,搅拌反应,得到含有蒽基的环氧化产物;
[0029]
步骤(2)、将步骤(1)得到的含有蒽基的环氧化产物溶于溶剂b中,加入 端巯基醇化合物和促进剂,搅拌反应,得到含有蒽基侧链的双端羟基化合物;
[0030]
步骤(3)、将双端异氰酸酯基化合物和双端羟基的聚己内酯溶于溶剂c中, 加入有机锡催化剂,搅拌反应,生成扩链的双端异氰酸酯基化合物;
[0031]
步骤(4)、将扩链的双端异氰酸酯基化合物与含有蒽基侧链的双端羟基化 合物和不含有蒽基侧链的双端羟基化合物溶于溶剂d中,加入有机锡催化剂, 搅拌反应,制得所述高强度光可逆胶粘剂(含有蒽侧基和聚己内酯结晶链段的 线型聚氨酯结构的化合物)。
[0032]
本发明的一种优选的实施方式中,
[0033]
步骤(1)中,
[0034]
所述蒽基化合物选自9
‑
蒽甲醇、1
‑
(9
‑
蒽基)乙醇中的至少一种;优选9
‑
蒽 甲醇;
[0035]
所述环氧化合物选自环氧基卤素化合物;优选环氧氯丙烷;
[0036]
所述碱选自氢氧化钠或氢氧化钾;优选氢氧化钠;
[0037]
所述相转移催化剂选自四甲胺卤素化合物;优选四甲基溴化铵;
[0038]
所述溶剂a为非极性溶剂;优选甲苯;
[0039]
蒽基化合物、环氧化合物、碱和相转移催化剂的摩尔比为1:(5~10):(2~4): (0.01~0.03);
[0040]
蒽基化合物和溶剂a的摩尔比为1:20~1:30;
[0041]
搅拌反应温度为60~70℃,搅拌反应时间为3~6h。
[0042]
本发明的一种优选的实施方式中,
[0043]
步骤(2)中,
[0044]
所述溶剂b为非极性溶剂;优选甲苯;
[0045]
所述端巯基醇化合物选自2
‑
巯基乙醇,3
‑
巯基
‑1‑
丙醇,4
‑
巯基
‑1‑
丁醇中的 至少一种;优选2
‑
巯基乙醇;
[0046]
所述促进剂选自叔胺类化合物;优选选用2、4、6
‑
三(二甲胺基甲基)苯酚 (dmp
‑
30);促进剂用量为含有蒽基的环氧化产物和端巯基醇化合物总重量的 0.5wt~1wt%;
[0047]
含有蒽基的环氧化物和溶剂b的摩尔比为1:20~1:30;
[0048]
含有蒽基的环氧化产物和端巯基醇化合物的摩尔比为1:1~1:1.1;
[0049]
搅拌反应温度为20~30℃,搅拌反应时间为2~3h。
[0050]
本发明的一种优选的实施方式中,
[0051]
步骤(3)中,
[0052]
所述双端异氰酸酯基化合物选自4,4
‑
二苯基甲烷二异氰酸酯,4,4
‑
二环 己基甲烷二异氰酸酯和六亚甲基二异氰酸酯中的一种;优选4,4
‑
二环己基甲烷 二异氰酸酯;
[0053]
双端羟基的聚己内酯的重均分子量为2000~4000,优选重均分子量2000的 原料;
[0054]
溶剂c为极性溶剂;优选甲基异丁基酮;
[0055]
所述有机锡催化剂为二月桂酸二丁基锡;催化剂用量为双端异氰酸酯基化 合物和双端羟基的聚己内酯总重量的0.5wt~1wt%;
[0056]
双端羟基的聚己内酯和溶剂c的摩尔比为1:30~1:40;
[0057]
双端异氰酸酯基化合物和双端羟基的聚己内酯的摩尔比为2:1~2.1:1;
[0058]
搅拌反应温度为70~80℃,搅拌反应时间为2~3h。
[0059]
本发明的一种优选的实施方式中,
[0060]
步骤(4)中,
[0061]
所述不含有蒽基侧链的双端羟基化合物选自1,4
‑
丁二醇,1,5
‑
戊二醇,1, 6
‑
己二醇等亚甲基二醇中的一种;优选1,4
‑
丁二醇;
[0062]
所述溶剂d为极性溶剂;优选甲基异丁基酮;
[0063]
异氰酸酯基与羟基的摩尔比为1:1~1.1:1;
[0064]
含有蒽基侧链的双端羟基化合物和不含有蒽基侧链的双端羟基化合物的摩 尔比为1:(0.25~4);
[0065]
所述有机锡催化剂为二月桂酸二丁基锡;催化剂用量为扩链双端异氰酸酯 基化合物、含有蒽基侧链的双端羟基化合物和不含有蒽基侧链的双端羟基化合 物总重量的0.5wt~1wt%;
[0066]
扩链双端异氰酸酯基化合物和溶剂d的摩尔比为1:30~1:40;
[0067]
搅拌反应温度为70~80℃,搅拌反应时间为2~3h。
[0068]
本发明的一种进一步优选的实施方式中,
[0069]
步骤(1)中得到的含蒽基的环氧化产物经离心、萃取、旋转蒸发和干燥; 其中,所述萃取溶剂选自甲苯、甲基异丁基酮,乙酸乙酯中的至少一种;
[0070]
上述离心、萃取、旋转蒸发和干燥等操作均可在任何可实现上述操作的常 用设备上来完成。
[0071]
步骤(2)、(3)、(4)中得到的产物在真空加热设备中除去溶剂,加热温度 为90~100℃,加热时间为2~3h。
[0072]
本发明的目的之三是提供一种所述方法制备的高强度光可逆胶粘剂。
[0073]
本发明的目的之四是提供一种高强度光可逆胶粘剂在透光性基材中的应 用。
[0074]
本发明的胶粘剂适用于各种透光性基材的粘接,尤其适用于石英玻璃的粘 接。
[0075]
与现有技术相比,本发明具有以下有益之处:
[0076]
1.本发明胶粘剂中的蒽基基团相对于其他光诱导可逆反应基团,如香豆素, 二硫键,具备更强的反应活性与可操作性;相对于光致异构化基团,如偶氮苯, 蒽基基团能够应用于热固性聚合物,具备稳定的交联结构,从而提供较强的粘 接强度与稳定性。
[0077]
2.本发明构造了一种半结晶性的光诱导可逆胶粘剂,结晶链段在低玻璃化 转变温度的交联网络的基础上提高了粘接强度与抗蠕变性;以无定形相的形式 存在的交联链段在较低的玻璃化转变温度下,依然能够确保可逆固化反应的顺 利进行。
[0078]
本发明提供的含有蒽基基团和聚己内酯结晶链段的高强度光可逆胶粘剂,由 于交联点的存在使得链段的排列受阻,因此交联链段基本以无定形相的形式存 在,在较低的玻璃化转变温度下,能够确保在350~405nm紫外光下诱导蒽基的 [4 4]环加成反应,从而发生交联,提供较强的粘接力;在紫外光波长(小于 300nm)时可以诱导环加成产物的解聚,发生解交联,使粘接力下降。光引发反 应具有快速反应的优势,能够使熔融体的链段在受到结晶作用的限制之前快速 固化成膜,紫外光光照时间为5~10min。同时,非交联链段则依然具备较强的结 晶能力,结晶链段的存在以及在低玻璃化转变温度体系的基础上,起到了增强 和抗蠕变的作用。本发明提供的光可逆胶粘剂粘接性能优异,拉伸剪切强度强 度可达3.8mpa,优于当前报道的其他光可逆胶粘剂;光解后交联结构丧失,重 新得到的线型结构可以轻易通过溶剂脱粘。脱粘过程中选用极性溶剂,本实验 中优先选用丙酮;溶剂处理时间为5~10min;经过溶剂处理后不再有聚合物残留 在基材上。
附图说明
[0079]
图1为实施例1中的9
‑
蒽甲醇、9
‑
蒽甲醇环氧化物和含有蒽基侧链的双端羟 基化合物的红外光谱图,其中曲线1a为9
‑
蒽甲醇(am)的红外光谱图,曲线1b为9
‑
蒽甲醇环氧化物(aer)的红外光谱图,曲线1c为含有蒽基侧链的双端 羟基化合物(aermt)的红外光谱图;
[0080]
图2为实施例1中得到的aer和aermt的1h nmr谱,其中曲线2a为 9
‑
蒽甲醇(am)的1h nmr谱,曲线2b为9
‑
蒽甲醇环氧化物(aer)的1h nmr 谱;
[0081]
图3为实施例1中得到的aer的
13
c nmr光谱;
[0082]
图4为实施例1中得到的aermt的
13
c nmr光谱;
[0083]
图5为实施例1中得到的扩链反应生成的大分子量的双端异氰酸酯基化合 物(pcl/hmdi)与含有蒽基侧链的双端羟基化合物(aermt)和1,4
‑
丁二醇 (bd)反应得到含有蒽侧基和聚己内酯结晶链段的线型聚氨酯结构的化合物 (apu);其中曲线5a为pcl/hmdi的红外光谱,曲线5b为apu的红外光谱;
[0084]
图6为采用xrd监测实施例1得到的apu的相态结构图;
[0085]
图7为采用gpc监测实施例1得到的apu分子在凝胶渗透色谱柱中的保 留时间图;
[0086]
图8为实施例1得到的apu的熔融体的dsc冷却
‑
加热扫描曲线,并且在 冷却过程中,在室温(25℃)下保温20min,表征室温下的结晶速率;
[0087]
图9为实施例1得到的apu的熔融体的粘
‑
温曲线;
[0088]
图10采用紫外可见分光光度计追踪实施例1得到的apu的光固化过程, 光照时间共5分钟;
[0089]
图11为实施例1得到的apu的结晶体的dsc升温扫描曲线(0~80℃), 其中曲线11a为固化前的apu升温扫描曲线,曲线11b为固化后的apu升温扫 描曲线;
[0090]
图12为实施例1得到的apu的结晶体的dsc升温扫描曲线(
‑
65~
‑
10℃), 其中曲线12a为固化前的apu升温扫描曲线,曲线12b为固化后的apu升温 扫描曲线;
[0091]
图13为实施例1得到的apu的拉伸剪切测试曲线,13a为实施例1得到的 apu交联并
且结晶后的拉伸剪切测试结果,13b为实施例1得到的apu交联并 且结晶后,又经过光诱导解交联的拉伸剪切测试结果。
[0092]
图14为实施例2中的9
‑
蒽甲醇、9
‑
蒽甲醇环氧化物和含有蒽基侧链的双端 羟基化合物的红外光谱图,其中曲线14a为9
‑
蒽甲醇(am)的红外光谱图,曲 线14b为9
‑
蒽甲醇环氧化物(aer)的红外光谱图,曲线14c为含有蒽基侧链 的双端羟基化合物(aermt)的红外光谱图;
[0093]
图15为实施例2中得到的aer和aermt的1h nmr谱,其中曲线15a 为9
‑
蒽甲醇(am)的1h nmr谱,曲线15b为9
‑
蒽甲醇环氧化物(aer)的1h nmr谱;
[0094]
图16为实施例2中得到的aer的
13
c nmr光谱;
[0095]
图17为实施例2中得到的aermt的
13
c nmr光谱;
[0096]
图18为实施例2中得到的扩链反应生成的大分子量的双端异氰酸酯基化合 物(pcl/hmdi)与含有蒽基侧链的双端羟基化合物(aermt)和1,4
‑
丁二醇(bd)反应得到含有蒽侧基和聚己内酯结晶链段的线型聚氨酯结构的化合物 (apu);其中曲线18a为pcl/hmdi的红外光谱,曲线18b为apu的红外光 谱;
[0097]
图19为采用xrd监测实施例2得到的apu的相态结构图;
[0098]
图20为采用gpc监测实施例2得到的apu分子在凝胶渗透色谱柱中的保 留时间图;
[0099]
图21为实施例2得到的apu的熔融体的dsc冷却
‑
加热扫描曲线,并且在 冷却过程中,在室温(25℃)下保温20min,表征室温下的结晶速率;
[0100]
图22为实施例2得到的apu的熔融体的粘
‑
温曲线;
[0101]
图23采用紫外可见分光光度计追踪实施例2得到的apu的光固化过程, 光照时间共5分钟;
[0102]
图24为实施例2得到的apu的结晶体的dsc升温扫描曲线(0~80℃), 其中曲线24a为固化前的apu升温扫描曲线,曲线24b为固化后的apu升温 扫描曲线;
[0103]
图25为实施例2得到的apu的结晶体的dsc升温扫描曲线(
‑
65~
‑
10℃), 其中曲线25a为固化前的apu升温扫描曲线,曲线25b为固化后的apu升温 扫描曲线;
[0104]
图26为实施例2得到的apu的拉伸剪切测试曲线,26a为实施例2得到的 apu交联并且结晶后的拉伸剪切测试结果,26b为实施例2得到的apu交联并 且结晶后,又经过光诱导解交联的拉伸剪切测试结果。
[0105]
图27为实施例3中的9
‑
蒽甲醇、9
‑
蒽甲醇环氧化物和含有蒽基侧链的双端 羟基化合物的红外光谱图,其中曲线27a为9
‑
蒽甲醇(am)的红外光谱图,曲 线27b为9
‑
蒽甲醇环氧化物(aer)的红外光谱图,曲线27c为含有蒽基侧链 的双端羟基化合物(aermt)的红外光谱图;
[0106]
图28为实施例3中得到的aer和aermt的1h nmr谱,其中曲线28a 为9
‑
蒽甲醇(am)的1h nmr谱,曲线28b为9
‑
蒽甲醇环氧化物(aer)的1h nmr谱;
[0107]
图29为实施例3中得到的aer的
13
c nmr光谱;
[0108]
图30为实施例3中得到的aermt的
13
c nmr光谱;
[0109]
图31为实施例3中得到的扩链反应生成的大分子量的双端异氰酸酯基化合 物(pcl/hmdi)与含有蒽基侧链的双端羟基化合物(aermt)和1,4
‑
丁二醇 (bd)反应得到含有蒽侧基和聚己内酯结晶链段的线型聚氨酯结构的化合物 (apu);其中曲线31a为pcl/hmdi
的红外光谱,曲线31b为apu的红外光 谱;
[0110]
图32为采用xrd监测实施例3得到的apu的相态结构图;
[0111]
图33为采用gpc监测实施例3得到的apu分子在凝胶渗透色谱柱中的保 留时间图;
[0112]
图34为实施例3得到的apu的熔融体的dsc冷却
‑
加热扫描曲线,并且在 冷却过程中,在室温(25℃)下保温20min,表征室温下的结晶速率;
[0113]
图35为实施例3得到的apu的熔融体的粘
‑
温曲线;
[0114]
图36采用紫外可见分光光度计追踪实施例3得到的apu的光固化过程, 光照时间共10分钟;
[0115]
图37为实施例3得到的apu的结晶体的dsc升温扫描曲线(0~80℃), 其中曲线37a为固化前的apu升温扫描曲线,曲线37b为固化后的apu升温 扫描曲线;
[0116]
图38为实施例3得到的apu的结晶体的dsc升温扫描曲线(
‑
65~
‑
10℃), 其中曲线38a为固化前的apu升温扫描曲线,曲线38b为固化后的apu升温 扫描曲线;
[0117]
图39为实施例3得到的apu的拉伸剪切测试曲线,39a为实施例3得到的 apu交联并且结晶后的拉伸剪切测试结果,39b为实施例3得到的apu交联并 且结晶后,又经过光诱导解交联的拉伸剪切测试结果。
具体实施方式
[0118]
下面结合具体附图及实施例对本发明进行具体的描述,有必要在此指出的 是以下实施例只用于对本发明的进一步说明,不能理解为对本发明保护范围的 限制,本领域技术人员根据本发明内容对本发明做出的一些非本质的改进和调 整仍属本发明的保护范围。
[0119]
实施例中所采用的测试仪器及测试条件如下:
[0120]
ftir:bruker alpha ftir,采用的是kbr压片法,并在4000~400cm
‑1的波 数范围内以4cm
‑1的分辨率上进行。
[0121]
nmr:核磁共振光谱法(1h
‑
nmr和
13
c
‑
nmr)在室温下使用cdcl3作为 溶剂在bruker av400mhz nmr光谱仪(德国)上进行。
[0122]
xrd:样品结晶区与非晶区的衍射峰在室温下,采用40kv,40ma,cu kα 辐射的x射线衍射仪(荷兰)采集。
[0123]
粘度:在25
‑
150℃温度范围内,加热速率为4℃/min,剪切速率为50s
‑1, 使用流变仪(mcr
‑
301,anton paar)和平行板工具测量粘度。
[0124]
gpc:采用waters 1525仪器,配备waters 2414ri检测器,四氢呋喃作为 流动相(流速:0.6ml/min;列温度:35℃)。
[0125]
uv
‑
vis:通过shimadzu uv
‑
2550光谱仪在吸收模式下进行uv
‑
vis光谱测试。 采用的是固态样品测试方法。
[0126]
dsc:在配备有rcs 90冷却系统的ta instruments q20上,在n2气氛(50 ml
·
min
‑1)下,对原始、解交联和再交联的网络进行dsc测试。所有样品均以 10℃/min的加热速率从
‑
50℃加热到100℃。
[0127]
拉伸剪切测试:拉伸剪切强度测试是在室温下通过sans utm5205xhd测 试设备进行的。采用单搭边剪切测试方法,粘接基材包括石英玻璃和pe膜。样 品架的移动速度设置
为10mm/min。
[0128]
实施例中所采用化合物来源如下:
[0129]
药品名称生产厂家9
‑
蒽甲醇安耐吉试剂有限公司环氧氯丙烷天津福辰(中国)化学有限公司氢氧化钠北京化工厂(中国)四甲基溴化铵北京化工厂(中国)甲苯天津福辰(中国)化学有限公司甲基异丁基酮天津福辰(中国)化学有限公司2
‑
巯基乙醇上海麦克林生化科技有限公司2、4、6
‑
三(二甲胺基甲基)苯酚美国圣地亚哥bioruler聚己内酯二醇天津希恩思生化科技有限公司二月桂酸二丁基锡上海麦克林生化科技有限公司1,4
‑
丁二醇天津福辰(中国)化学有限公司4,4
‑
二环己基甲烷二异氰酸酯上海麦克林生化科技有限公司蒸馏水自制石英玻璃片中国连云港威达石英有限公司
[0130]
实施例1
[0131]
胶粘剂结构式:
[0132][0133]
0<q<8;0<p<7;0<n<8;0<m<6;且p,q,n,m为整数; 其中,
[0134]
a为
[0135][0136]
b为:
[0137][0138]
a=19
[0139]
d为:
[0140][0141]
b=4
[0142]
r为
[0143][0144]
制备方法包括:
[0145]
1)含蒽的环氧化产物的制备:将1mol 9
‑
蒽甲醇(am)原料溶于25mol非 极性溶剂甲苯中,再加入8mol环氧氯丙烷,水浴保持温度在65℃,然后加入 3mol的氢氧化钠和0.02mol的四甲基溴化铵(tmab)。保温搅拌反应4h,通过 离心、甲苯萃取、旋转蒸发和真空干燥以获得含蒽基基团的环氧化产物——9
‑ꢀ
蒽甲醇环氧化物(aer);
[0146]
2)含有蒽基侧链的双端羟基化合物的制备:步骤1)制得的9
‑
蒽甲醇环氧化 物0.5mol与0.525mol巯基乙醇混合,溶于12.5mol甲苯,以质量分数的0.75% 加入dmp
‑
30促进剂,搅拌均匀后在25℃下反应2.5h,通过旋转蒸发和真空干 燥以获得含蒽基基团的环氧化产物(aermt);
[0147]
3)扩链的双端异氰酸酯基化合物的制备:0.041mol 4,4
‑
二环己基甲烷二异 氰酸酯(hmdi)和0.02mol双端羟基的聚己内酯(m
w
=2000),即聚己内酯二醇 (pcl
‑
2oh),溶于0.7mol甲基异丁基酮溶剂中,以质量分数的0.75%加入二月 桂酸二丁基锡(dbtdl)催化剂,搅拌均匀后在75℃反应2.5h,通过旋转蒸发 和真空干燥以获得扩链的双端异氰酸酯基化合物(pcl/hmdi);
[0148]
4)含有蒽侧基和聚己内酯结晶链段的线型聚氨酯结构的化合物的制备:将 0.021mol pcl/hmdi与0.01mol的aermt和0.01mol的1,4
‑
丁二醇(bd)溶 于0.7mol甲基异丁基酮溶剂中,异氰酸酯基与羟基的摩尔比为1.05:1,以质量 分数的0.75%加入二月桂酸二丁基锡(dbtdl)催化剂,搅拌均匀后在75℃反 应2.5h,得到高强度光可逆胶粘剂(apu
‑
1)。
[0149]
5)固化:步骤4)制备得到的胶粘剂预先在60℃下预热1min使其熔融,用 于固定两片石英玻璃基材,施加额定的预压强(20kpa),随后将熔融体转移至 365nm波长紫外光(40w)下照射5min使其固化,得到固化后的含有蒽基基团 和聚己内酯结晶链段的胶粘剂(cl
‑
apu
‑
1)。
[0150]
对得到的固化后的胶粘剂置于室温(25℃)下2h使其充分结晶,并进行拉 伸剪切测试,测得对石英玻璃的粘接强度为3.8mpa(图13a);在254nm紫外光 (8w)下照射2h,胶粘剂光降解后的强度为2.1mpa(图13b),由于结晶链段 仍然存在,胶粘剂依然具备一定的粘接强度。将光解后的胶粘剂于丙酮中浸泡 10分钟,光解后重新得到的线型聚氨酯聚合物在丙酮中溶解,并且不再有聚合 物残留在基材上。
[0151]
测试及表征:
[0152]
1.aer和aermt的红外光谱表征
[0153]
图1为实施例1中的9
‑
蒽甲醇(am)、9
‑
蒽甲醇环氧化物(aer)和含有 蒽基侧链的双端羟基化合物(aermt)的红外光谱图,图中,1a为am的红外 光谱图,在3400cm
‑1附近的宽峰归属于
‑
oh的振动,经过反应后,可以观察到 这些吸收消失,表明
‑
oh在反应中被消耗完全;1b为aer的光谱,在913cm
‑1处的吸收代表了环氧基团的特征吸收,表明环氧氯丙烷和
‑
oh基团之间的环氧 化反应已成功进行。1c为aermt的光谱,在913cm
‑1处的吸收峰消失,3400cm
‑1附近归属于
‑
oh的宽峰重新出现,证明环氧基团开环反应形成了
‑
oh。
[0154]
2.aer和aermt的nmr表征
[0155]
为了进一步确认结构,我们对aer和aermt进行了nmr表征。图2为 aer和aermt的1h nmr,图3和图4分别为aer和aermt的
13
c nmr 光谱,从谱图中可以清楚地观察到,nmr谱图中的峰与产物的质子与c原子都 一一对应,以上结果可以充分证明已成功合成aer和aermt。
[0156]
3.apu
‑
1的红外表征
[0157]
图5表征了扩链的双端异氰酸酯基化合物(pcl/hmdi)与含有蒽基侧链的 双端羟基化合物(aermt)和1,4
‑
丁二醇(bd)反应生成含有蒽侧基和聚己 内酯结晶链段的线型聚氨酯结构的胶粘剂(apu
‑
1)的过程。图5a的曲线代表 pcl/hmdi的光谱,图5b的曲线代表apu
‑
1的光谱;在2260cm
‑1左右的吸收 峰代表
‑
nco,3300cm
‑1左右的吸收峰代表
‑
n
‑
h,apu的光谱图中2260cm
‑1左右 的吸收峰消失,而3300cm
‑1左右的吸收峰相对高度变大,证明
‑
oh与
‑
nco发生 反应消耗了
‑
nco,生成了
‑
n
‑
h。
[0158]
4.apu
‑
1的xrd表征
[0159]
图6通过xrd表征了apu
‑
1的相态结构,范围从10
°
到30
°
显示了一个经 典的非晶态宽频带,在21
°
和23
°
的晶体峰与聚己内酯二醇(pcl
‑
2oh)的晶体 链段相对应。这表明apu
‑
1体系中晶相与非晶相并存。
[0160]
5.apu
‑
1的可操作性表征
[0161]
通过图7的gpc测试来表征apu
‑
1产物的分子量与分子量分布,得到数均 分子量(m
n
)为16704,重均分子量(m
w
)为41382,分子量分布(pdi)为 2.48;图8通过dsc降温
‑
升温扫描表征了apu
‑
1室温下的结晶速率,通过公式 [0162]
计算了在室温下保温20min后的结晶度,其中δh
m
代表图8中的熔融焓值,δh
c
代表图8中的结晶焓值,二者相减得到的差值可以认为是在室温下保温过程中 的结晶焓值,δh
100%
代表pcl的理论结晶焓,通过查阅文献可知该值等于 135j/g,通过计算得到室温下保温20min后的结晶度为3.3%,由于光固化过程 在5min内完成,因此该产物具备足够的可操作性;如图9所示,通过流变仪测 试了apu
‑
1的粘
‑
温曲线,得到室温下熔融体的粘度为152930mpa.s。
[0163]
6.固化紫外可见吸收光谱追踪
[0164]
为了获得apu
‑
1最佳的固化时间以及确认固化完全,采用紫外可见吸收光 谱跟踪实施例1中步骤5的交联固化过程。用紫外灯(40w)在365nm的紫外 线下照射不同时长以得到不同固化程度的样品,如图10。从图中可以看出,图 中4个代表蒽基的吸收峰随着光照时间的延长逐渐降低,5min后吸收值不再变 化。通过公式:
[0165][0166]
计算了固化反应程度,a0表示光照之前390nm波长处的吸收值,a1表示光照之 后390nm波长处的吸收值,经计算得到固化率达到了89%。
[0167]
7.apu
‑
1固化前后结晶度与玻璃化转变温度
[0168]
dsc可以用来监测apu
‑
1固化前后结晶度与玻璃化转变温度的变化,如图 11和图12,图11中a和b分别为实施例1中固化前与固化后的结晶熔融曲线, 图12中的a和b分别为实施例1中固化前与固化后的玻璃化转变温度曲线。通 过公式:
[0169][0170]
其中δh
m
代表曲线11a和曲线11b中的熔融焓值,δh
100%
代表pcl的理论结晶 焓,通过查阅文献可知该值等于135j/g,经计算得到固化前的结晶度为30.3%, 固化后的结晶度为20.8%,由于交联结构限制了链段的运动,使得固化后的结晶 链段下降。曲线12a没有表现出玻璃化转变温度,这是由于固化前玻璃化转变 温度太低超出了dsc的测试量程,曲线12b表明固化后的玻璃化转变温度为
ꢀ‑
47.9℃。
[0171]
8.apu
‑
1固化后与光解后的粘接强度表征
[0172]
将apu
‑
1熔融体涂覆在石英基板上,并将另一片石英基板覆盖其上,用 20kpa恒定的压强使胶粘剂充分浸润粘接接头,粘接面积为75mm2。随后,用 365nm的紫外光(20w)照射5分钟使其固化,并在室温下静止2h使其充分结 晶。利用搭接剪切强度测试方法测试了粘接强度,其粘接强度可达到3.8mpa, 如图13a所示,优于当前报道的其他光可逆胶粘剂。
[0173]
使用与以上相同方式制备的搭接样品,在254nm紫外光下(8w)照射1h, 依旧通过搭接剪切强度测试方法测试了粘接强度,发现粘接强度显著下降至 2.1mpa,如图13b所示,但是由于结晶结构依然存在,粘接接头依旧具备一定 的粘接强度,将光解后的胶粘剂于丙酮中浸泡10分钟,光解后重新得到的线型 聚氨酯聚合物在丙酮中溶解,并且不再有聚合物残留在基材上。
[0174]
实施例2
[0175]
胶粘剂的结构式同实施例1.
[0176]
制备方法包括:
[0177]
1)含蒽的环氧化产物的制备:将1mol 9
‑
蒽甲醇(am)原料溶20mol非 极性溶剂甲苯中,再加入5mol环氧氯丙烷,水浴保持温度在60℃,然后加入 2mol的氢氧化钠和0.01mol的四甲基溴化铵(tmab)。保温搅拌反应6h,通过 离心、甲苯萃取、旋转蒸发和真空干燥以获得含蒽基基团的环氧化产物——9
‑ꢀ
蒽甲醇环氧化物(aer);
[0178]
2)含有蒽基侧链的双端羟基化合物的制备:步骤1)制得的9
‑
蒽甲醇环氧 化物0.5mol与0.5mol巯基乙醇混合,溶于10mol甲苯,以质量分数的0.5%加 入dmp
‑
30促进剂,搅拌均匀后在室温(20℃)下反应3h,通过旋转蒸发和真 空干燥以获得含蒽基基团的环氧化产物(aermt);
[0179]
3)扩链的双端异氰酸酯基化合物的制备:0.04mol 4,4
‑
二环己基甲烷二 异氰酸酯(hmdi)和0.02mol双端羟基的聚己内酯(m
w
=2000),即聚己内酯二 醇(pcl
‑
2oh),溶于0.6mol甲基异丁基酮溶剂中,以质量分数的0.5%加入二 月桂酸二丁基锡(dbtdl)催化剂,搅拌均匀后在70℃反应3h,通过旋转蒸发 和真空干燥以获得扩链的双端异氰酸酯基化合物(pcl/hmdi);
[0180]
4)含有蒽侧基和聚己内酯结晶链段的线型聚氨酯结构的化合物的制备: 将0.02mol pcl/hmdi与0.016mol的aermt和0.004mol的1,4
‑
丁二醇(bd) 溶于0.6mol甲基异丁基酮溶剂中,异氰酸酯基与羟基的摩尔比为1:1,以质量分 数的0.5%加入二月桂酸二丁基锡(dbtdl)催化剂,搅拌均匀后在70℃反应 3h,得到高强度光可逆胶粘剂(apu
‑
2)。
[0181]
5)固化:步骤4)制备得到的胶粘剂预先在60℃下预热1min使其熔融, 用于固定两片石英玻璃基材,施加额定的预压强(20kpa),随后将熔融体转移 至405nm波长紫外光
(40w)下照射5min使其固化,得到固化后的含有蒽基基 团和聚己内酯结晶链段的高强度光可逆胶粘剂(cl
‑
apu
‑
2)。
[0182]
对得到的固化后的胶粘剂置于室温(25℃)下2h使其充分结晶,并进行拉 伸剪切测试,测得对石英玻璃的粘接强度为3.1mpa,在254nm紫外光(8w) 下照射2h,光降解后的强度为1.7mpa,由于结晶链段仍然存在,胶粘剂依然具 备一定的粘接强度。将光解后的胶粘剂于丙酮中浸泡10分钟,光解后重新得到 的线型聚氨酯聚合物在丙酮中溶解,并且不再有聚合物残留在基材上。
[0183]
测试及表征:
[0184]
1.aer和aermt的红外光谱表征
[0185]
图14为实施例2中的9
‑
蒽甲醇(am)、9
‑
蒽甲醇环氧化物(aer)和含有 蒽基侧链的双端羟基化合物(aermt)的红外光谱图,图中,14a为am的红 外光谱图,在3400cm
‑1附近的宽峰归属于
‑
oh的振动,经过反应后,可以观察 到这些吸收消失,表明
‑
oh在反应中被消耗完全;14b为aer的光谱,在913cm
‑1处的吸收代表了环氧基团的特征吸收,表明环氧氯丙烷和
‑
oh基团之间的环氧 化反应已成功进行。14c为aermt的光谱,在913cm
‑1处的吸收峰消失,3400cm
‑1附近归属于
‑
oh的宽峰重新出现,证明环氧基团开环反应形成了
‑
oh。
[0186]
2.aer和aermt的nmr表征
[0187]
为了进一步确认结构,我们对aer和aermt进行了nmr表征。图15为 aer和aermt的1h nmr,图16和图17分别为aer和aermt的
13
c nmr 光谱,从谱图中可以清楚地观察到,nmr谱图中的峰与产物的质子与c原子都 一一对应,以上结果可以充分证明已成功合成aer和aermt。
[0188]
3.apu
‑
2的红外表征
[0189]
图18表征了扩链的双端异氰酸酯基化合物(pcl/hmdi)与含有蒽基侧链 的双端羟基化合物(aermt)和1,4
‑
丁二醇(bd)反应生成含有蒽侧基和聚 己内酯结晶链段的线型聚氨酯结构的化合物(apu
‑
2)的过程。图18a的曲线代 表pcl/hmdi的光谱,图18b的曲线代表apu
‑
2的光谱;在2260cm
‑1左右的吸 收峰代表
‑
nco,3300cm
‑1左右的吸收峰代表
‑
n
‑
h,apu
‑
2的光谱图中2260cm
‑1左右的吸收峰消失,而3300cm
‑1左右的吸收峰相对高度变大,证明
‑
oh与
‑
nco 发生反应消耗了
‑
nco,生成了
‑
n
‑
h。
[0190]
4.apu
‑
2的xrd表征
[0191]
图19通过xrd表征了apu
‑
2的相态结构,范围从10
°
到30
°
显示了一个经 典的非晶态宽频带,在21
°
和23
°
的晶体峰与聚己内酯二醇(pcl
‑
2oh)的晶体 链段相对应。这表明apu
‑
2体系中晶相与非晶相并存。
[0192]
5.apu
‑
2的可操作性表征
[0193]
通过图20的gpc测试来表征apu
‑
2产物的分子量与分子量分布,得到数 均分子量(m
n
)为14598,重均分子量(m
w
)为38902,分子量分布(pdi)为 2.66;图21通过dsc降温
‑
升温扫描表征了apu
‑
2室温下的结晶速率,通过公 式计算了在室温下保温20min后的结晶度,其中δh
m
代表图21中的熔融焓值,δh
c
代表图21中的结晶焓值,二者相减得到的差值可以认为是在室温下保温过程中 的结晶焓值,δh
100%
代表pcl的理论结晶焓,通过查阅文献可知该值等于 135j/g,通过计算得到室温下保温20min后的结晶度
为2.0%,由于光固化过程 在5min内完成,因此该产物具备足够的可操作性;因为相对于产物apu
‑
1来说, apu
‑
2具有较多的侧基,分子链间的距离增大,使得等温条件下的结晶速率变 慢,结晶度也降低。如图22所示,通过流变仪测试了apu
‑
2的粘
‑
温曲线,得 到室温下熔融体的粘度为280520mpa.s。与apu
‑
1相比,在相对分子量与分子 量分布接近的情况下,两种样品的黏度有着明显的区别,原因可能是较多的芳 香侧基直接有较强的π
‑
π相互作用。
[0194]
6.固化紫外可见吸收光谱追踪
[0195]
为了获得apu
‑
2最佳的固化时间以及确认固化完全,采用紫外可见吸收光 谱跟踪实施例2中步骤5的交联固化过程。用紫外灯(40w)在405nm的紫外 线下照射不同时长以得到不同固化程度的样品,如图23。从图中可以看出,图 中4个代表蒽基的吸收峰随着光照时间的延长逐渐降低,5min后吸收值不再变 化。通过公式:
[0196][0197]
计算了固化反应程度,a0表示光照之前390nm波长处的吸收值,a1表示光照之 后390nm波长处的吸收值,经计算得到固化率达到了75%。这是由于样品apu
‑
2 较大的黏度对于链段运动有一定限制,使得固化率相对apu
‑
1降低。
[0198]
7.apu
‑
2固化前后结晶度与玻璃化转变温度
[0199]
dsc可以用来监测apu
‑
2固化前后结晶度与玻璃化转变温度的变化,如图 24和图25,图24中a和b分别为实施例2中固化前与固化后的结晶熔融曲线, 图25中的a和b分别为实施例2中固化前与固化后的玻璃化转变温度曲线。通 过公式:
[0200][0201]
其中δh
m
代表曲线24a和曲线24b中的熔融焓值,δh
100%
代表pcl的理论结晶 焓,通过查阅文献可知该值等于135j/g,经计算得到固化前的结晶度为24.6%, 固化后的结晶度为18.9%,由于交联结构限制了链段的运动,使得固化后的结晶 链段下降。而依据前文中的描述,apu
‑
2无论在固化前还是固化后,结晶度都 应低于apu
‑
1,这是正常的。曲线25a没有表现出玻璃化转变温度,这是由于 固化前玻璃化转变温度太低超出了dsc的测试量程,曲线25b表明固化后的玻 璃化转变温度为
‑
54.5℃。虽然理论上讲,apu
‑
2的交联密度要大于apu
‑
1,但 是其玻璃化转变温度却低于apu
‑
1,这一现象也是由于样品apu
‑
2的结晶度相 比于apu
‑
1来说较低导致的。
[0202]
8.apu
‑
2固化后与光解后的粘接强度表征
[0203]
将apu
‑
2熔融体涂覆在石英基板上,并将另一片石英基板覆盖其上,用 20kpa恒定的压强使胶粘剂充分浸润粘接接头,粘接面积为75mm2。随后,用 365nm的紫外光(20w)照射5分钟使其固化,并在室温下静止2h使其充分结 晶。利用搭接剪切强度测试方法测试了粘接强度,其粘接强度可达到3.1mpa, 如图26a所示,优于当前报道的其他光可逆胶粘剂。通过数据可以看出,实施 例2中样品的粘接强度低于实施例1,这是由于样品的粘接强度是由交联结构与 结晶结构共同保持的,二者的协同作用起到了高粘接强度的效果。因此,对于 交联链段与结晶链段的比例调控是非常重要的。其中,实施例1中样品的比例 明显是更加合适的,而实施列2中的样品结晶链段含量较少,对于粘接力的增 强作用稍弱。
[0204]
使用与以上相同方式制备的搭接样品,在254nm紫外光下(8w)照射1h, 依旧通过,
搭接剪切强度测试方法测试了粘接强度,发现粘接强度显著下降至 1.7mpa,如图26b所示,但是由于结晶结构依然存在,粘接接头依旧具备一定 的粘接强度,将光解后的胶粘剂于丙酮中浸泡10分钟,光解后重新得到的线型 聚氨酯聚合物在丙酮中溶解,并且不再有聚合物残留在基材上。
[0205]
实施例3
[0206]
胶粘剂结构式同实施例1.
[0207]
制备方法包括:
[0208]
1)含蒽的环氧化产物的制备:将1mol 9
‑
蒽甲醇(am)原料溶于30mol 非极性溶剂甲苯中,再加入10mol环氧氯丙烷,水浴保持温度在70℃,然后加 入4mol的氢氧化钠和0.03mol的四甲基溴化铵(tmab)。保温搅拌反应6h, 通过离心、甲苯萃取、旋转蒸发和真空干燥以获得含蒽基基团的环氧化产物 ——9
‑
蒽甲醇环氧化物(aer);
[0209]
2)含有蒽基侧链的双端羟基化合物的制备:步骤1)制得的9
‑
蒽甲醇环氧 化物0.5mol与0.55mol巯基乙醇混合,溶于15mol甲苯,以质量分数的1%加入 dmp
‑
30促进剂,搅拌均匀后在室温(30℃)下反应2h,通过旋转蒸发和真空 干燥以获得含蒽基基团的环氧化产物(aermt);
[0210]
3)扩链的双端异氰酸酯基化合物的制备:0.042mol 4,4
‑
二环己基甲烷二 异氰酸酯(hmdi)和0.02mol双端羟基的聚己内酯(m
w
=2000),即聚己内酯二 醇(pcl
‑
2oh),溶于0.8mol甲基异丁基酮溶剂中,以质量分数的1%加入二月 桂酸二丁基锡(dbtdl)催化剂,搅拌均匀后在80℃反应2h,通过旋转蒸发和 真空干燥以获得扩链的双端异氰酸酯基化合物(pcl/hmdi);
[0211]
4)含有蒽侧基和聚己内酯结晶链段的线型聚氨酯结构的化合物的制备: 将0.022mol pcl/hmdi与0.004mol的aermt和0.016mol的1,4
‑
丁二醇(bd) 溶于0.8mol甲基异丁基酮溶剂中,异氰酸酯基与羟基的摩尔比为1.1:1,以质量 分数的1%加入二月桂酸二丁基锡(dbtdl)催化剂,搅拌均匀后在80℃反应 2h,得到高强度光可逆胶粘剂(apu
‑
3)。
[0212]
5)固化:步骤4)制备得到的胶粘剂预先在60℃下预热1min使其熔融, 用于固定两片石英玻璃基材,施加额定的预压强(20kpa),随后将熔融体转移 至405nm波长紫外光(40w)下照射10min使其固化,得到固化后的含有蒽基 基团和聚己内酯结晶链段的高强度光可逆胶粘剂(cl
‑
apu
‑
3)。
[0213]
固化后的胶粘剂置于室温(25℃)下2h使其充分结晶,并进行拉伸剪切测 试,测得对石英玻璃的粘接强度为3.0mpa(图31a),在254nm紫外光(8w) 下照射2h,光降解后的强度为2.8mpa(图31b),由于结晶链段仍然存在,胶粘 剂依然具备一定的粘接强度。将光解后的胶粘剂于丙酮中浸泡30分钟,光解后 重新得到的线型聚氨酯聚合物在丙酮中溶解,并且不再有聚合物残留在基材上。
[0214]
测试及表征:
[0215]
1.aer和aermt的红外光谱表征
[0216]
图27为实施例3中的9
‑
蒽甲醇(am)、9
‑
蒽甲醇环氧化物(aer)和含有 蒽基侧链的双端羟基化合物(aermt)的红外光谱图,图中,27a为am的红 外光谱图,在3400cm
‑1附近的宽峰归属于
‑
oh的振动,经过反应后,可以观察 到这些吸收消失,表明
‑
oh在反应中被消耗完全;27b为aer的光谱,在913cm
‑1处的吸收代表了环氧基团的特征吸收,表明环氧氯丙烷和
‑
oh基团之间的环氧 化反应已成功进行。27c为aermt的光谱,在913cm
‑1处的吸收峰消失,3400cm
‑1附近归属于
‑
oh的宽峰重新出现,证明环氧基团开环反应形成了
‑
oh。
[0217]
2.aer和aermt的nmr表征
[0218]
为了进一步确认结构,我们对aer和aermt进行了nmr表征。图28为 aer和aermt的1h nmr,图29和图30分别为aer和aermt的
13
c nmr 光谱,从谱图中可以清楚地观察到,nmr谱图中的峰与产物的质子与c原子都 一一对应,以上结果可以充分证明已成功合成aer和aermt。
[0219]
3.apu
‑
3的红外表征
[0220]
图31表征了扩链的双端异氰酸酯基化合物(pcl/hmdi)与含有蒽基侧链 的双端羟基化合物(aermt)和1,4
‑
丁二醇(bd)反应生成含有蒽侧基和聚 己内酯结晶链段的线型聚氨酯结构的化合物(apu
‑
3)的过程。图31a的曲线代 表pcl/hmdi的光谱,图31b的曲线代表apu
‑
3的光谱;在2260cm
‑1左右的吸 收峰代表
‑
nco,3300cm
‑1左右的吸收峰代表
‑
n
‑
h,apu
‑
3的光谱图中2260cm
‑1左右的吸收峰消失,而3300cm
‑1左右的吸收峰相对高度变大,证明
‑
oh与
‑
nco 发生反应消耗了
‑
nco,生成了
‑
n
‑
h。
[0221]
4.apu
‑
3的xrd表征
[0222]
图32通过xrd表征了apu
‑
3的相态结构,范围从10
°
到30
°
显示了一个经 典的非晶态宽频带,在21
°
和23
°
的晶体峰与聚己内酯二醇(pcl
‑
2oh)的晶体 链段相对应。这表明apu
‑
3体系中晶相与非晶相并存。
[0223]
5.apu
‑
3的可操作性表征
[0224]
通过图33的gpc测试来表征apu
‑
3产物的分子量与分子量分布,得到数 均分子量(m
n
)为11957,重均分子量(m
w
)为25609,分子量分布(pdi)为 2.14;图34通过dsc降温
‑
升温扫描表征了apu
‑
3室温下的结晶速率,通过公 式计算了在室温下保温20min后的结晶度,其中δh
m
代表图34中的熔融焓值,δh
c
代表图34中的结晶焓值,二者相减得到的差值可以认为是在室温下保温过程中 的结晶焓值,δh
100%
代表pcl的理论结晶焓,通过查阅文献可知该值等于 135j/g,通过计算得到室温下保温20min后的结晶度为14.3%。结晶速率较快是 因为其中芳香侧基含量减少,分子链段排布更为密集。这使得样品apu
‑
3的可 操作性低于apu
‑
1与apu
‑
2。如图35所示,通过流变仪测试了apu
‑
3的粘
‑
温 曲线,得到室温下熔融体的粘度为338950mpa.s,相对于apu
‑
1与apu
‑
2来说, apu
‑
3的相对分子量更小,但是却具备更高的黏度,原因是因为较高的结晶度 与结晶速率导致的。综合三组apu样品的测试结果来看,芳香侧基的含量过多 与过少都会对结晶性、黏度以至于可操作性产生不利影响。
[0225]
6.固化紫外可见吸收光谱追踪
[0226]
为了获得apu
‑
3最佳的固化时间以及确认固化完全,采用紫外可见吸收光 谱跟踪实施例3中步骤5的交联固化过程。用紫外灯(40w)在365nm的紫外 线下照射不同时长以得到不同固化程度的样品,如图36。从图中可以看出,图 中4个代表蒽基的吸收峰随着光照时间的延长逐渐降低,10min后吸收值不再变 化。通过公式:
[0227]
[0228]
计算了固化反应程度,a0表示光照之前390nm波长处的吸收值,a1表示光照之 后390nm波长处的吸收值,经计算得到固化率达到了70%。综合前文所述,这 也是由于黏度较高的影响导致的。
[0229]
7.apu
‑
3固化前后结晶度与玻璃化转变温度
[0230]
dsc可以用来监测apu
‑
3固化前后结晶度与玻璃化转变温度的变化,如图 37和图38,图37中a和b分别为实施例3中固化前与固化后的结晶熔融曲线, 图38中的a和b分别为实施例3中固化前与固化后的玻璃化转变温度曲线。通 过公式:
[0231][0232]
其中δh
m
代表曲线37a和曲线37b中的熔融焓值,δh
100%
代表pcl的理论结晶 焓,通过查阅文献可知该值等于135j/g,经计算得到固化前的结晶度为30.3%, 固化后的结晶度为20.8%,由于交联结构限制了链段的运动,使得固化后的结晶 链段下降。曲线38a没有表现出玻璃化转变温度,这是由于固化前玻璃化转变 温度太低超出了dsc的测试量程,曲线38b表明固化后的玻璃化转变温度为
ꢀ‑
35.9℃。虽然理论上讲,apu
‑
1和apu
‑
2的交联密度要大于apu
‑
3,但是其玻 璃化转变温度却都低于apu
‑
3,这一现象也是由于样品apu
‑
3的结晶度较高导 致的。
[0233]
8.apu
‑
3固化后与光解后的粘接强度表征
[0234]
将apu
‑
3熔融体涂覆在石英基板上,并将另一片石英基板覆盖其上,用 20kpa恒定的压强使胶粘剂充分浸润粘接接头,粘接面积为75mm2。随后,用 365nm的紫外光(20w)照射5分钟使其固化,并在室温下静止2h使其充分结 晶。利用搭接剪切强度测试方法测试了粘接强度,其粘接强度可达到3.0mpa, 如图39a所示,优于当前报道的其他光可逆胶粘剂。通过数据可以看出,实施 例3中样品的粘接强度也要低于实施例1。这是由于实施例3中的样品交联结构 较少,这与实施例2一样,同样说明了样品的粘接强度是由交联结构与结晶结 构共同保持的,二者的协同作用起到了高粘接强度的效果,对于二者之间比例 的调节是非常重要的。
[0235]
使用与以上相同方式制备的搭接样品,在254nm紫外光下(8w)照射1h, 依旧通过,搭接剪切强度测试方法测试了粘接强度,发现粘接强度显著下降至 2.8mpa。我们发现,相对于实施例1和2,实施例3的强度下降幅度最小。这是 因为相比于apu
‑
1与apu
‑
2样品来说,apu
‑
3样品交联密度最小,结晶度最高, 相对应的,经过解交联反应后剪切强度降低的也最少。如图39b所示,由于结 晶结构依然存在,粘接接头依旧具备一定的粘接强度,将光解后的胶粘剂于丙 酮中浸泡30分钟,光解后重新得到的线型聚氨酯聚合物在丙酮中溶解,并且不 再有聚合物残留在基材上。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。