一种氘代吡咯烷酮衍生物、药物组合物及其用途
发明领域
1.本发明属于医药技术领域,具体涉及氘代吡咯烷酮衍生物、药物 组合物及其用途。更具体地说,本发明涉及所述的化合物制备的方法、 包含其的药物组合物及使用其激活葡萄糖激酶作为治疗ⅱ型糖尿病 药物。
背景技术:
2.糖尿病(diabetes mellitus,dm)是由于体内胰岛素绝对不足 或相对不足所引起的以慢性高血糖为主要特征,合并脂肪和蛋白质代 谢紊乱的综合征。随着糖尿病病程的延长,患者易并发心、脑、肾、 视网膜及神经系统慢性进行性病变。糖尿病引发多种并发症,严重影 响患者的生命健康和生活质量。长期的血糖升高导致糖尿病患者微血 管损伤,并引起外周神经损伤、视网膜病变、糖尿病肾病等。
[0003]ⅱ型糖尿病又称为非胰岛素依赖型糖尿病,是由组织细胞的胰岛 素抵抗、胰岛β细胞功能衰退和其他多种原因引起的。在糖尿病患 者中,ⅱ型糖尿病约占90%。目前,临床用于治疗ⅱ型糖尿病的降糖 药物种类繁多,机制各异,但都无法阻止胰岛β细胞功能进行性衰 退,病程进展难以逆转。因此,临床上急需寻找一种既可以使血糖达 标,又能保护甚至修复残存胰岛β细胞的药物。
[0004]
葡萄糖激酶(glucokinase,gk)是一种己糖激酶,主要分布在 成熟肝细胞和胰岛β细胞中,参与葡萄糖代谢的第一步反应,催化 葡萄糖转变为葡萄糖
‑6‑
磷酸进入肝脏和胰腺,发挥重要的“葡萄糖 传感器”的作用,对于人体血糖稳态的调控意义重大(ma y, ratnasabapathy r,izzi
‑
engbeaya c,et al.hypothalamic arcuateglucokinase regulates insulin secretion and glucose homeostasis[j].diabetes obes metab,2018,20(9): 2246
‑
2254.doi:10.1111/dom.13359)。葡糖激酶激活剂 (glucokinase activator,gka)是针对葡萄糖激酶这个靶点而开发 的新型化合物,能够通过葡萄糖浓度刺激胰岛素分泌,降低胰高血糖 素浓度和肝糖输出,促进肝糖原合成以及调控肠促胰素释放来稳定体 内血糖水平(余刚,葡萄糖激活剂研究进展[j],药学进展,2016 年3月,第40卷,第3期,168
‑
177页)。
[0005]
自2003年gka进入研究以来,gka的研究不断深入,各制药公 司和研究机构都争相加入该研究领域。目前,以吡咯烷酮为基本结构 的gka前景最为乐观,其代表化合物hms5552已进入临床ⅲ期研究。 研究表明,hms5552是一种拥有全新作用机制的t2dm治疗药物,能 够激活人体gk的功能,提高人体对葡萄糖的敏感性,改善胰岛素分 泌;同时,可提高肝糖原合成的效率,使得肝脏、肌肉和脂肪对葡萄 糖的摄取及储存能力显著提高(胡玉玺,陈永收,任一鑫,曹爽,治 疗2型糖尿病的仔研新药—葡萄糖激酶激动剂hms5552[j],临床药 物治疗杂志,2020年6月,第18卷,第6期,1
‑
5页)。简而言之, hms5552通过激活肝脏和胰脏中的gk达到降糖控糖的目的。
[0006]
与传统的降糖药相比,gka以全新的作用机制为ⅱ型糖尿病提供 了新的治疗方法。然而,关于gka转化成为药物的研究仅仅只是开端, 此领域还需要进一步开发对葡萄糖
激酶有激动作用或更好药效学/药 代动力学性能的化合物。
技术实现要素:
[0007]
本发明提供一种含氘代吡咯烷酮类化合物及其衍生物的葡萄糖激 酶激活剂,具有较好的药代动力学性能。一种式i所示的氘代吡咯烷 酮衍生物、其立体异构体、互变异构体、水合物、溶剂合物、活性代 谢物、多晶体、共晶体、药学上可接受的盐及其前药;
[0008][0009]
其中:
[0010]
r1、r2、r3、r4独立地选自氢,氘;
[0011]
r5可以为氢或者氘;
[0012]
r6、r7独立地选自氢,氘;
[0013]
r8、r9、r
10
独立地选自氢,氘;
[0014]
r
11
、r
12
、r
13
独立地选自氢,氘;
[0015]
r
14
可以为氢或者氘;
[0016]
r
15
、r
16
独立地选自氢,氘;
[0017]
r
17
可以为氢或者氘;
[0018]
r
18
、r
19
独立地选自氢,氘;
[0019]
且r1、r2、r3、r4、r5、r6、r7、r8、r9、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、 r
16
、r
17
、r
18
、r
19
中至少一个是氘。
[0020]
当r5为氘原子时,其优选结构如下式ii:
[0021][0022]
其中r6、r7、r8、r9、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、 r
19
独立的选自氢或者氘。
[0023]
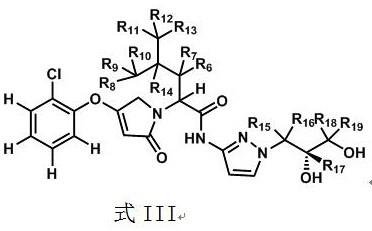
当r5为氢原子时,其优选结构如下式iii:
[0024][0025]
其中r6、r7、r8、r9、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
独立的选自氢或氘,并且这其中至少有一个是氘。
[0026]
如通式i所示的氘代吡咯烷酮衍生物、其立体异构体、互变异构 体、水合物、溶剂合物、活性代谢物、多晶体、共晶体、药学上可接 受的盐及其前药,其中所述的化合物优先选自以下化合物:
[0027][0028]
在其它方面中,本发明还提供药物组合物,其包含药学上可 接受的载体和本发明的化合物;治疗或预防病症或病况的方 法,所述病症或病况选自以下的代谢紊乱、减缓该代谢紊乱的进 展、或治疗该代谢紊乱:i型糖尿病、ii型糖尿病、葡萄糖耐量降低、 高血糖症、餐后高血糖症、空腹血糖异常、超重、肥胖症、高血压症、 胰岛素抵抗和/或代谢综合征;或改善血糖控制和/或降低空腹血浆葡 萄糖;或预防、减缓、延迟或逆转糖尿病并发症。
[0029]
本发明中提供的含氘代吡咯烷酮类化合物及其衍生物的葡萄糖 激酶激活剂及可以与其组合其它药剂包括治疗ii型糖尿病的药物: glp
‑
1受体激动剂,艾塞那肽、利拉鲁肽、索马鲁肽、度拉鲁肽、洛 塞那肽、阿必鲁肽;α
‑
葡萄糖苷酶抑制剂,伏格列波糖、阿卡波糖; sglt
‑
2抑制剂,坎格列净(canagliflozin)、达格列净 (dapagliflozin)、恩格列净
(empagliflozin)、依格列净 (ipragliflozin)、鲁格列净(luseogliflozin)以及托格列净 (tofogliflozin);dpp
‑
4抑制剂,西格列汀(sitagliptin)、维格列 汀(vildagliptin)、沙格列汀(saxagliptin)、阿格列汀(alogliptin)、 利格列汀(linagliptin)、吉格列汀(gemigliptin)和替格列汀 (teneligliptin)和二甲双胍中的一种或多种。其药物组合物可用 于预防一种或多种选自以下的代谢紊乱、减缓该代谢紊乱的进展、或 治疗该代谢紊乱:i型糖尿病、ii型糖尿病、葡萄糖耐量降低、高血 糖症、餐后高血糖症、空腹血糖异常、超重、肥胖症、高血压症、胰 岛素抵抗和/或代谢综合征;或改善血糖控制和/或降低空腹血浆葡萄 糖;或预防、减缓、延迟或逆转糖尿病并发症。
[0030]
本发明化合物或其药学上可接受的盐的纯净形式或适当药物组 合物可通过任何可接受的投予起类似效用的药剂的模式投予。本发明 药物组合物可通过将本发明化合物与适当药学上可接受的载剂、稀释 剂或赋形剂组合而制成,并且可调配成固体、半固体、液体或气体形 式制剂,例如片剂、胶囊、散剂、颗粒剂、溶液、注射剂、吸入剂、 凝胶、微球体和气雾剂。投予所述药物组合物的典型途径包括(但不 限于)经口、局部、透皮、吸入、不经肠、舌下、颊、直肠、和鼻 内投药。如本文中所使用,术语不经肠包括皮下注射、静脉内、肌内、 胸骨内注射或输注技术。本发明药物组合物经调配,以允许在对患者 投予组合物后,其中所含活性成分是生物可利用的。将被投予个体或 患者的组合物里一种或多种剂量单位的形式,其中,例如片剂可为单 剂量单位,而含气雾剂形式的本发明化合物的容器可容纳多个剂量单 位。制备所述剂型的实际方法为所属领域技术人员已知,或将为其所 知悉。欲投予的组合物在任何情况下都将含有治疗有效量的本发明化 合物或其药学上可接受的盐,以便根据本发明的示教治疗所关注的疾 病或病状。
[0031]
本发明药物组合物可呈固体或液体形式。一方面,载剂为微粒, 以致组合物例如呈片剂或散剂形式。载剂可为液体,而组合物为例如 口服糖浆、可注射液体,或适用于例如吸入投药的气雾剂。当欲口服 时,药物组合物优选自固体或液体形式,其中半固体、半液体、悬 浮液和凝胶形式包括在本文中视为固体或液体的形式中。对于口服固 体组合物,可将药物组合物调配成散剂、颗粒剂、压缩片剂、丸剂、 胶囊、咀嚼片、粉片等形式。此类固体组合物通常含有一种或多种惰 性稀释剂或可食用载剂。此外,还可存在一种或多种以下物质粘合剂, 例如羧甲基纤维素、乙基纤维素、微晶纤维素、黄瓦胶或明胶; 赋形剂,例如淀粉、乳糖或糊精;崩解剂,例如海藻酸、海藻酸纳、 primogel、玉米淀粉等;润滑剂,例如硬脂酸镁或氢化植物油 (sterotex);助流剂,例如胶状二氧化硅;甜味剂,例如蔗糖或糖精; 调味剂,例如薄荷、水杨酸甲酌或甜橙调味剂;和着色剂。
[0032]
药物组合物为胶囊形式,例如为明胶胶囊时,除上述类型的物质 以外,其还可含有液体载剂,例如聚乙二醇或植物油。药物组合物可 为液体形式,例如酊剂、糖浆、溶液、乳液或悬浮液。此液体可口服, 或通过注射递送,作为两个实例。当欲口服时,优选组合物除含有本 发明化合物以外,还含有甜味剂、防腐剂、染料/着色剂和风味增强 剂中的一种或多种。在打算通过注射投予的组合物中,可包括表面活 性剂、防腐剂、润湿剂、分散剂、悬浮剂、缓冲剂、稳定剂和等渗剂 中的一种或多种。
具体实施方式
[0033]
下面通过实施例的方式进一步说明本发明,提供本技术中所述的 合成实施例和
生物学实施例以说明本文所提供的化合物、药物组合物、 以及方法。以下实施例仅用于对本发明进行示例性说明,但不用于限 制本发明,在本发明保护范围内所做的修改、改变、变型等都在本发 明的保护范围内。
[0034]
本文所提供的化合物可以使用下文所阐述的特定合成方案的将为 本领域技术人员公知的操作方案,由容易获得的起始物质来制备。下 列实施例中未注明具体条件的实验方法,按照常规方法和条件,可以 由本领域技术人员通过常规的优化程序来确定。
[0035]
下述实施例中,缩写解释:
[0036]
pe:石油醚;
[0037]
ea:乙酸乙酯;
[0038]
dmf:n,n
‑
二甲基甲酰胺;
[0039]
dcm:二氯甲烷;
[0040]
thf:四氢呋喃
[0041]
meoh:甲醇
[0042]
boc2o:二叔丁基二碳酸酯;
[0043]
diea:n,n
‑
二异丙基乙胺
[0044]
dmso:二甲基亚砜
[0045]
nbs:n
‑
溴代丁二酰亚胺
[0046]
ccl4:四氯化碳
[0047]
tea:三乙胺
[0048]
dabco:三乙烯二胺
[0049]
toscl:4
‑
甲基苯磺酰氯
[0050]
h
‑
leu
‑
ome.hcl:l
‑
亮氨酸甲酯盐酸盐
[0051]1h nmr:核磁共振氢谱
[0052]
tlc:薄层层析
[0053]
chiral hplc:手性高效液相色谱
[0054]
prep
‑
hplc:高压制备液相色谱
[0055]
lc
‑
ms:液相色谱
‑
质谱联用
[0056]
rf:比移值;
[0057]
min:分钟
[0058]
g;克
[0059]
mg:毫克
[0060]
rt:室温
[0061]
mol:摩尔
[0062]
mmol:毫摩尔
[0063]
ml:毫升
[0064]
m:摩尔/升
[0065]
实施例1:(s)
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑ꢀ
戊酸[1
‑
((r)
‑
2,3
‑
二羟基
‑
1,1,2,3,3
‑
五氘代
‑
丙基)
‑1‑
氢
‑
吡唑
‑3‑
基]
‑
酰胺 (化合物1)的制备:
[0066][0067][0068]
(e)
‑3‑
(2
‑
氯
‑
苯氧基)
‑2‑
丁烯酸乙酯(1c)的制备:将2
‑
氯苯酚(1a) (10.9ml,107mmol)溶解于400ml mecn中,依次加入dabco(9.8 g,87.4mmol)、2
‑
丁炔酸乙酯(1b)(10.4ml,89.2mmol),反应混 合物加热至70℃搅拌3小时。反应混合物冷却至室温,用二氯甲烷 和水萃取2次,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残 留物经柱层析纯化获得化合物1c(17.0g,产率:90%)。ms(esi):m/z 240.9[m 1]
[0069]
(e)
‑4‑
溴
‑3‑
(2
‑
氯
‑
苯氧基)
‑2‑
丁烯酸乙酯(1d)的制备:将化合 物1c(16.0g,66.7mmol)溶解于300ml四氯化碳中,依次加入 nbs(13.0g,87.4mmol)、过氧化苯甲酰(170mg,0.7mmol),反 应混合物加热至回流,反应过夜。反应混合物冷却至室温,过滤,滤 液减压浓缩,残留物经柱层析纯化获得化合物1d(21.0g,产率:99%)。 ms(esi):m/z 319.0[m 1]
,321.0[m 1]
(溴同位素).
[0070]
(s)
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸甲酯 (1e)的制备:在室温下,将化合物1d(3.2g,10mmol)和l
‑
亮氨 酸甲酯盐酸盐(1.45g,10mmol)溶解于50ml乙腈中,加入diea (3.9g,30mmol),反应混合物加热至80℃,反应过夜。反应混合 物冷却至室温,过滤,滤液减压浓缩,残留物经柱层析纯化获得化合 物1e(1.4g,产率:42%)。ms(esi):m/z 338.2[m]
[0071]
(s)
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸(1f) 的制备:将化合物1e(1.4g,4.1mmol)溶解于30ml甲醇中,加 入水30ml,反应混合物冷却至0℃。加入氢氧化锂(344mg,8.2 mmol),反应3小时。减压蒸去甲醇。用1m hcl调节ph至3,二 氯甲烷抽提2次。有机相用饱和食盐水洗涤2次,无水硫酸钠干燥, 过滤,滤液减压蒸干,得化合物1f(900mg,产率:67%)。ms(esi): m/z 322.2[m
‑
h]
‑
[0072]
2,2
‑
二甲基
‑4‑
(1,1
‑
二氘代)
‑
羟甲基
‑
4,5,5
‑
三氘代
‑
1,3
‑
二氧环戊烷(1h) 的制备:将1,1,2,3,3
‑
五氘代丙三醇(1g)(1.0g,10.3mmol)溶解于 150ml四氢呋喃中,依次加入2,2
‑
二甲氧基丙烷(4.3g,41.2mmol)、 磷钨酸(100mg),反应混合物在室温下搅拌过夜。tlc检测反应完 毕,减压蒸去溶剂,得粗产物1h(1.5g,产率:100%)直接用于下一 步反应。
[0073]4‑
甲基苯磺
‑
[1,1
‑
二氘代
‑1‑
(4,5,5
‑
三氘代
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑ꢀ
基)]甲酯(1i)的制备:将化合物1h(1.4g,10.3mmol)溶解于100 ml二氯甲烷中,加入tea(4.2g,41.2mmol),反应混合物冷却至 0℃。加入toscl(3.9g,20.6mmol),加毕,撤去冰浴,室温下搅拌 3小时。tlc检测反应完毕后,加入适量水,二氯甲烷抽提3次。有 机相用饱和食盐水洗涤2次,无水硫酸钠干燥,过滤,滤液减压蒸干, 经柱层析纯化获得化合物1i(1.8g,产率:60%)。
[0074]
n
‑
[1,1
‑
二氘代
‑1‑
(4,5,5
‑
三氘代
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基)甲 基]
‑
1h
‑
吡唑
‑3‑
胺(1k)的制备:将化合物1j(500mg,6mmol)溶 解于10ml dmso中,加入koh(1.0g,18mmol),反应混合物冷 却至0℃。加入化合物1i(1.8g,6mmol),加毕,撤去冰浴,室温 下反应过夜。tlc检测反应完毕,加入适量水,二氯甲烷抽提3次。 有机相用饱和食盐水洗涤2次,无水硫酸钠干燥,过滤,滤液减压蒸 干,经柱层析纯化获得化合物1k(490mg,产率:41%)。
[0075]
n
‑
[1,1
‑
二氘代
‑1‑
((r)
‑
4,5,5
‑
三氘代
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基)甲 基]
‑
1h
‑
吡唑
‑3‑
胺(1l)的制备:将化合物1k(490mg)经chiral hplc 分离后获得化合物1l(220mg,产率:90%)。
[0076]
(s)
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸[1,1
‑
二 氘代
‑1‑
((r)
‑
4,5,5
‑
三氘代
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基甲基
‑
1h
‑
吡唑
ꢀ‑3‑
基)]
‑
酰胺(1m)的制备:将化合物1f(150mg,0.46mmol)溶解 于10ml二氯甲烷中,依次加入edci(178mg,0.93mmol)和hobt (125mg,0.93mmol),室温下搅拌30min。加入化合物1l(112mg, 0.56mmol),室温下反应过夜。tlc检测反应毕,加入适量水,二氯 甲烷抽提3次。有机相用饱和食盐水洗涤2次,无水硫酸钠干燥,过 滤,滤液减压蒸干,经柱层析纯化获得化合物1m(210mg,产率:89%)。 (s)
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸 [1
‑
((r)
‑
2,3
‑
二羟基
‑
1,1,2,3,3
‑
五氘代
‑
丙基)
‑
h
‑
吡唑
‑3‑
基]
‑
酰胺(1)的 制备:将化合物1m(210mg,0.41mmol)溶解于5ml乙腈中,加 入水1ml和磷钨酸30mg,混合物在36℃下搅拌2小时。lc
‑
ms检 测反应完毕,经prep
‑
hplc(acetonitrile/h2o 0.1%nh3h2o)获得化 合物1(101mg,产率:52%)。1h nmr(400mhz,cdc
l3
):δ(ppm):9.7 (br,1h),7.49
‑
7.47(m,1h),7.36
‑
7.34(m,1h),7.32
‑
7.30(m,1h), 7.29
‑
7.22(m,2h),6.64(d,j=2.4hz,1h),4.88
‑
4.81(m,2h),4.45(d,j =18.4hz,1h),4.15(d,j=18.0hz,1h),1.87
‑
1.72(m,2h),1.58
‑
1.51 (m,1h),0.99
‑
0.94(m,6h);hplc purity:99.8%(214nm),99.9%(254 nm);ms(esi):m/z 468.2[m h]
;氘代率97.4%
[0077]
实施例2:(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢
‑
吡咯
‑1‑ꢀ
基]
‑4‑
甲基
‑
戊酸[1
‑
((r)
‑
2,3
‑
二羟基
‑
丙基)
‑
1h
‑
吡唑
‑3‑
基]
‑
酰胺(化合物 2)的制备
[0078][0079]2‑
氘代
‑2‑
氨基
‑4‑
甲基戊酸(2b)的制备:将化合物2a(5.0g,38mmol) 溶解于50ml ch3cood中,加入水杨醛(0.5ml),混合物加热至 110℃,反应2小时。冷却至室温,减压蒸去溶剂后,重新加入 ch3cood(50ml)和水杨醛(0.5ml),混合物加热至110℃,反 应2小时。重复4次这项操作,减压蒸去溶剂,加入d2o(100ml), 室温下搅拌30min,过滤,滤液减压浓缩,获得黄色油状物2b(4.1g, 粗产物)。
[0080]2‑
氘代
‑2‑
氨基
‑4‑
甲基戊酸甲酯(2c)的制备:将化合物2b(4.1g, 粗产物,31mmol)溶解于50ml无水甲醇中,冷却至0℃,加入socl
2 (7.3g,62mmol),加毕,撤去冰浴,室温下反应过夜。减压蒸去溶 剂,加入适量水,饱和nahco3调节ph至8,乙酸乙酯抽提3次。 有机相用饱和食盐水洗涤2次,无水硫酸钠干燥,过滤,滤液减压蒸 干,得黄色油状物2c(1.6g,粗产物)。
[0081]2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢
‑
吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸甲 酯(2d)的制备:将化合物1d(3.8g,12.1mmol)和化合物2c(1.6 g,11mmol)溶解于50ml乙腈中,加入diea(4.3g,33mmol), 反应混合物加热至80℃,反应过夜。反应混合物冷却至室温,过滤, 滤液减压浓缩,残留物经柱层析纯化获得化合物2d(600mg,产率: 16%)。1h nmr(400mhz,cdcl3):δ(ppm):7.49
‑
7.47(m,1h), 7.34
‑
7.30(m,1h),7.24
‑
7.21(m,1h),4.85(s,1h),4.42(d,j=17.6hz, 1h),4.04(d,j=17.6hz,1h),3.72(s,3h),1.78
‑
1.70(m,2h),1.53
‑
1.50 (m,1h),0.98(t,j=6.8hz,6h).
[0082]2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢
‑
吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸 (2e)的制备:
[0083]
将化合物2d(600mg,1.8mmol)溶解于30ml甲醇中,加入水30 ml,反应混合物冷却至0℃。加入氢氧化锂(151mg,3.6mmol), 反应3小时。减压蒸去甲醇。用1m hcl调节ph至3,二氯甲烷抽 提2次。有机相用饱和食盐水洗涤2次,无水硫酸钠干燥,过滤,滤 液减压蒸干,得化合物2e(390mg,产率:67%)。
[0084]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢
‑
吡咯
‑1‑
基]
‑4‑
甲基
‑
戊 酸(2f)的制备:将化合物2e(390mg)经chiral hplc分离后获得 化合物2f(100mg,产率:51%)。1h nmr(400mhz,cdcl3):δ(ppm): 7.50
‑
7.48(m,1h),7.34
‑
7.30(m,1h),7.25
‑
7.22(m,2h),4.88(s,1h), 4.39(d,j=17.6hz,1h),4.04(d,j=17.6hz,1h),1.85
‑
1.72(m,2h), 1.60
‑
1.56(m,1h),0.98(t,j=6.4hz,6h);hplc purity:99.63% (214nm),99.82%(254nm);ms(esi):m/z 325.1[m h]
[0085]
n
‑
(1h
‑
吡唑
‑3‑
基)
‑
乙酰胺(2h)的制备:将化合物1j(9.0g,108.3mmol) 溶解于120ml乙酸乙酯中,室温下搅拌30min,缓慢加入ac2o (10.23ml,108.3mmol),混合物加热至60℃,搅拌6小时。冷却 至室温,过滤,滤饼用适量乙酸乙酯洗涤,真空干燥,得化合物2h (13.5g,产率:87%)。ms(esi):m/z 125.1[m 1]
[0086]
n
‑
[1
‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基甲基)
‑
1h
‑
吡唑
‑3‑
基]乙酰胺 (2j)的制备:将化合物2h(6.0g,48mmol)溶解于100ml dmf中, 依次加入叔丁醇钠(9.2g,96mmol)和氯化锂(2.0g,48mmol), 室温下搅拌30min。加入化合物2i(8.0g,53mmol),混合物加热 至100℃反应6小时。冷却至室温,加入适量水,二氯甲烷抽提3次。 有机相用饱和食盐水洗涤2次,无水硫酸钠干燥,过滤,滤液减压蒸 干,经柱层析纯化获得化合物2j(4.5g,产率:41%)。
[0087]1‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基甲基)
‑
1h
‑
吡唑
‑3‑
胺(2k)的制 备:将化合物2j(4.5g,18.8mmol)溶解于100ml水中,混合物冷 却至0℃。缓慢加入氢氧化钠(1.5g,37.6mmol),加毕,升温至90℃ 反应过夜。加入适量水,二氯甲烷抽提2次。有机相用饱和食盐水洗 涤2次,无水硫酸钠干燥,过滤,滤液减压蒸干,经柱层析纯化获得 化合物2k(3.6g,产率:97%)。
[0088]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸 [1
‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基甲基)
‑
1h
‑
吡唑
‑3‑
基]
‑
酰胺(2l) 的制备:将化合物2f(100mg,0.31mmol)溶解于10ml二氯甲烷中, 依次加入edci(118mg,0.62mmol)和hobt(83mg,0.62mmol), 室温下搅拌30min。加入化合物2k(91mg,0.46mmol),室温下反 应过夜。tlc检测反应完毕后,加入适量水,二氯甲烷抽提3次。有 机相用饱和食盐水洗涤2次,
无水硫酸钠干燥,过滤,滤液减压蒸干, 经柱层析纯化获得化合物2l(140mg,产率:90%)。
[0089]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸[1
‑
((r)
‑
2,3
‑
二羟基
‑
丙基)
‑
1h
‑
吡唑
‑3‑
基]
‑
酰胺(2)的制备:将化合物 2l(140mg,0.28mmol)溶解于5ml乙腈中,加入水1ml和磷钨 酸30mg,混合物在36℃下搅拌2小时。lc
‑
ms检测反应完毕,经 prep
‑
hplc(acetonitrile/h2o 0.1%nh3h2o)获得化合物2(72mg,产 率:56%)。1h nmr(400mhz,cdcl3):δ(ppm):7.49
‑
7.47(m,1h), 7.40(br,1h),7.35
‑
7.30(m,1h),7.29
‑
7.22(m,2h),6.63(s,1h),4.88(s, 1h),4.47(d,j=17.6hz,1h),4.24
‑
4.13(m,4h),3.69
‑
3.65(m,1h), 3.60
‑
3.56(m,1h),1.87
‑
1.72(m,2h),1.57
‑
1.53(m,1h),0.98(t,j=6.8 hz,6h);hplc purity:99.5%(214nm),99.8%(254nm);ms(esi):m/z 464.1[m h]
;氘代率96.5%
[0090][0091]
实施例3:(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢
‑
吡咯
‑1‑ꢀ
基]
‑4‑
甲基
‑
戊酸[1
‑
((r)
‑
2,3
‑
二羟基
‑
1,1,2,3,3
‑
五氘代
‑
丙基)
‑1‑
氢
‑
吡唑
ꢀ‑3‑
基]
‑
酰胺(化合物3)的制备:
[0092]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸 [1,1
‑
二氘代
‑1‑
((r)
‑
4,5,5
‑
三氘代
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基甲基
ꢀ‑
1h
‑
吡唑
‑3‑
基)]
‑
酰胺(3a)的制备:将化合物2f(120mg,0.37mmol) 溶解于10ml二氯甲烷中,依次加入edci(141mg,0.74mmol) 和hobt(100mg,0.74mmol),室温下搅拌30min。加入化合物1f (112mg,0.56mmol),室温下反应过夜。tlc检测反应完毕,加入 适量水,二氯甲烷抽提3次。有机相用饱和食盐水洗涤2次,无水硫 酸钠干燥,过滤,滤液减压蒸干,经柱层析纯化获得化合物3a(100 mg产率:53%)。
[0093]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸 [1
‑
((r)
‑
2,3
‑
二羟基
‑
1,1,2,3,3
‑
五氘代
‑
丙基)
‑
h
‑
吡唑
‑3‑
基]
‑
酰胺(3)的 制备:将化合物3a(100mg,0.2mmol)溶解于5ml乙腈中,加入 水1ml和磷钨酸30mg,混合物在36℃下搅拌2小时。lc
‑
ms检测 反应完毕,经prep
‑
hplc(acetonitrile/h2o 0.1%nh3h2o)获得化合 物3(74mg,产率:80%)。1h nmr(400mhz,cdcl3):δ(ppm):9.7(br, 1h),7.49
‑
7.47(m,1h),7.36
‑
7.34(m,1h),7.32
‑
7.30(m,1h),7.29
‑
7.22 (m,2h),6.64(d,j=2.4hz,1h),4.86(s,1h),4.45(d,j=18.4hz,1h), 4.15(d,j=18.4hz,1h),1.87
‑
1.72(m,2h),1.58
‑
1.51(m,1h),0.99
‑
0.94 (m,6h);hplc purity:99.9%(214nm),100%(254nm);ms(esi):m/z 469.2
[m h]
;氘代率97.3%
[0094]
实施例4:(s)
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢
‑
吡咯
‑1‑
基]
‑4‑
甲基
‑ꢀ
戊酸[1
‑
((r)
‑
1,1
–
二氘代
‑
2,3
‑
二羟基
‑
丙基)
‑1‑
氢
‑
吡唑
‑3‑
基]
‑
酰胺(化 合物4)的制备
[0095][0096]
1,1
‑
二氘代
‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑
基)甲醇(4b)的制备:在无 水无氧条件下,将化合物4a(0.5ml,3.46mmol)溶解于18ml无 水四氢呋喃中,加入ch3od(5.3ml),反应混合物冷却至0℃。硼 氘化钠(1.0g,23.9mmol)分3次加入。加毕,撤去冰浴,室温下 反应过夜。往反应混合物加入冰水(0.5ml),待没有气泡冒出后再 加入水(20ml)和甲基叔丁基醚(20ml)。二氯甲烷抽提3次,有 机相用饱和食盐水洗涤2次。无水硫酸钠干燥,过滤,滤液减压蒸干, 得淡黄色液体4b(396mg,粗产物)。
[0097]4‑
氯
‑
苯磺[1,1
‑
二氘代
‑1‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑
基)甲酯(4d) 的制备:将化合物4b(396mg,2.95mmol)溶解于6.5ml乙酸乙酯 中,加入dabco(414mg,3.69mmol),反应混合物冷却至0℃。加 入4
‑
氯苯磺酰氯(4c)(654mg,3.1mmol),保持0℃反应3小时, 撤去冰浴,室温下反应过夜。往反应混合物加入冰水(3ml),搅拌 15min。加入水(17ml)和乙酸乙酯(10ml),分出有机相,水相 用乙酸乙酯抽提2次。合并有机相,用饱和食盐水洗涤2次,无水硫 酸钠干燥,过滤,滤液减压蒸干,经柱层析纯化获得化合物4d(665 mg,产率:62.7%,两步合计)。1h nmr(400mhz,cdcl3):δ(ppm):7.87 (d,j=7.2hz,2h),7.54(d,j=7.2hz,2h),4.28(t,j=4.8hz,1h), 4.05(m,1h),3.77(m,1h),13.4(s,3h),1.31(s,3h).
[0098]
n
‑
[(1,1
‑
二氘代
‑1‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基)甲基)
‑
1h
‑
吡唑
ꢀ‑3‑
基]乙酰胺(4e)的制备:将化合物2i(250mg,2mmol)溶解于5 ml dmf中,加入叔丁醇钠
(231mg,2.4mmol),室温下搅拌30min。 加入化合物4d(741mg,2.4mmol),混合物加热至80℃,反应过夜。 冷却至室温,加入适量水,二氯甲烷抽提3次。有机相用饱和食盐水 洗涤2次,无水硫酸钠干燥,过滤,滤液减压蒸干,经柱层析纯化获 得化合物4e(383mg,产率:80%)。ms(esi):m/z 242.2[m h]
,264.2 [m na]
[0099]
[1,1
‑
二氘代
‑1‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基)甲基]
‑
1h
‑
吡唑
‑3‑ꢀ
胺(4f)的制备:将化合物4e(920mg,3.81mmol)溶解于4ml甲 醇中,加入水(4ml),混合物冷却至0℃。缓慢加入氢氧化钠(611 mg,15.25mmol),加毕,升温至90℃反应过夜。加入适量水,乙酸 乙酯抽提3次。有机相用饱和食盐水洗涤2次,无水硫酸钠干燥,过 滤,滤液减压蒸干,得化合物4f(620mg,产率:81.7%)。
[0100]
ms(esi):m/z 200.3[m h]
[0101]
(s)
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸[1,1
‑
二 氘代
‑1‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基甲基)
‑
1h
‑
吡唑
‑3‑
基]
‑
酰胺 (4g)的制备:将化合物1f(178mg,0.55mmol)溶解于5.5ml二 氯甲烷中,依次加入edci(116mg,0.61mmol)和hobt(82mg, 0.61mmol),室温下搅拌30min。加入化合物4f(131mg,0.66mmol) 和tea(61.2mg,0.61mmol),室温下反应过夜。lc
‑
ms检测反应 完毕,加入适量水,二氯甲烷抽提3次。有机相用饱和食盐水洗涤2 次,无水硫酸钠干燥,过滤,滤液减压蒸干,经柱层析纯化获得化合 物4g(103mg,产率:37%)。ms(esi):m/z 505.0[m h]
[0102]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸 [1
‑
((r)
‑
2,3
‑
二羟基
‑
丙基)
‑
1h
‑
吡唑
‑3‑
基]
‑
酰胺(4)的制备:将化合物 4g(103mg,0.2mmol)溶解于0.4ml异丙醇中,加入2m hcl 0.4ml, 混合物在室温下搅拌2小时。lc
‑
ms检测反应完毕,加入水(4ml) 和甲基叔丁基醚(10ml),分出有机相,水相用甲基叔丁基醚抽提2 次。合并有机相,依次用1m naoh溶液和饱和食盐水洗涤,无水硫 酸钠干燥,过滤,滤液减压蒸干,经dcm
‑
pe沉淀,获得化合物4 (84mg,产率:89%)。
[0103]1h nmr(400mhz,cdcl3):δ(ppm):9.68(s,1h),7.48
‑
7.46 (m,1h),7.32
‑
7.29(m,2h),7.24
‑
7.21(m,2h),6.63(s,1h), 4.87
‑
4.85(m,2h),4.46(d,j=14.4hz,1h),4.14(d,j=14.4hz, 1h),4.05(t,j=4.0hz,1h),3.66
‑
3.63(m,1h),3.54
‑
3.51(m,1h), 1.85
‑
1.79(m,1h),1.77
‑
1.71(m,1h),1.57
‑
1.51(m,1h),0.97(d,j =5.2hz,3h),0.94(d,j=5.2hz,3h);hplc purity:97.3% (214nm),98.0%(254nm);ms(esi):m/z 465.2[m h]
;氘代率 99%
[0104]
实施例5:(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢
‑
吡咯
‑1‑ꢀ
基]
‑4‑
甲基
‑
戊酸[1
‑
((r)
‑
2,3
‑
二羟基
‑
1,1,2,3,3
‑
五氘代
‑
丙基)
‑1‑
氢
‑
吡唑
ꢀ‑3‑
基]
‑
酰胺(化合物1)的制备
[0105][0106]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊 酸[1,1
‑
二氘代
‑1‑
((r)
‑
2,2
‑
二甲基
‑
1,3
‑
二氧戊环
‑4‑
基甲基)
‑
1h
‑
吡唑
‑3‑ꢀ
基]
‑
酰胺(5a)的制备:将化合物2f(40mg,0.123mmol)溶解于1.6 ml二氯甲烷中,依次加入edci(35.4mg,0.185mmol)和hobt (25mg,0.185mmol),室温下搅拌20min。加入化合物4f(37mg, 0.185mmol)和tea(18.7mg,0.185mmol),室温下反应过夜。tlc 检测反应完毕,加入适量水,二氯甲烷抽提3次。有机相用饱和食盐 水洗涤2次,无水硫酸钠干燥,过滤,滤液减压蒸干,经柱层析纯化 获得化合物5a(35mg,产率:56%)。
[0107]
(s)
‑2‑
氘代
‑2‑
[4
‑
(2
‑
氯
‑
苯氧基)
‑2‑
氧代
‑
2,5
‑
二氢吡咯
‑1‑
基]
‑4‑
甲基
‑
戊酸 [1
‑
((r)
‑
2,3
‑
二羟基
‑
丙基)
‑
1h
‑
吡唑
‑3‑
基]
‑
酰胺(5)的制备:将化合物 4g(35mg,0.2mmol)溶解于0.3ml异丙醇中,加入2m hcl 0.3ml, 混合物在室温下搅拌2小时。lc
‑
ms检测反应完毕,加入水(4ml) 和甲基叔丁基醚(10ml),分出有机相,水相用甲基叔丁基醚抽提2 次。合并有机相,依次用1m naoh溶液和饱和食盐水洗涤,无水硫 酸钠干燥,过滤,滤液减压蒸干,经dcm
‑
pe沉淀,获得化合物4 (26mg,产率:80.6%)。1h nmr(400mhz,cdcl3):δ(ppm):9.70(s, 1h),7.49
‑
7.46(m,1h),7.33
‑
7.28(m,2h),7.25
‑
7.21(m,2h),6.62(s, 1h),4.88
‑
4.85(m,2h),4.46(d,j=14.4hz,1h),4.14(d,j=14.4hz, 1h),3.67
‑
3.64(m,1h),3.53
‑
3.50(m,1h),1.85
‑
1.80(m,1h),1.77
‑
1.70 (m,1h),1.58
‑
1.51(m,1h),0.98(d,j=5.2hz,3h),0.94(d,j=5.2hz, 3h);hplc purity:95.3%(214nm),98.2%(254nm);ms(esi):m/z 466.0[m h]
;氘代率96.5%
[0108]
实施例6:
[0109]
体外葡萄糖激酶激动活性测试
[0110]
葡萄糖激酶(gck)实验步骤:
[0111]
1)准备100x梯度稀释的化合物:用dmso(sigma,d8418)将参 比化合物(mk
‑
0941(mce,hy
‑
19843))和待测化合物从1mm起3倍 稀释,每个化合物稀释10个浓度。
[0112]
2)准备3x梯度稀释化合物:在96孔稀释板(nunc,249944)中 加32.3ul的1x反应缓冲液,向每孔中转移1μl(1)中已梯度稀 释好的100x化合物。
[0113]
3)准备3x阳性对照(30μm mk
‑
0941)和3x阴性对照(3% dmso)稀释在1x反应缓冲液中。
[0114]
4)向384孔反应板(corning,3702)中加入6μl(2)和(3)中准 备好的化合物及阳性
对照和阴性对照。
[0115]
5)酶溶液的配制:用1x的反应缓冲液配制3x葡萄糖激酶 (recombinant human glucokinase/gck protein,r&d, 7840
‑
gk
‑
020)溶液。
[0116]
6)向(4)中的反应板每孔加入6μl 3x葡萄糖激酶稀释液。
[0117]
7)反应底物的配制:用1x的反应缓冲液配制3x的反应底物,使 其包含3x葡萄糖(d
‑
( )
‑
glucose,sigma,g5767),3xatp (atp/adenosine 5
′‑
triphosphate disodium salt hydrate, sigma,a7699),3x葡萄
‑6‑
磷酸脱氢酶(glucose
‑6‑
phosphatedehydrogenase,sigma,g6378)和3x nadp
(β
‑
nicotinamideadenine dinucleotide phosphate hydrate,sigma,n5755
‑
100mg)。
[0118]
8)向(6)中的反应板中每孔加入6μl的3x反应底物。
[0119]
9)使用victor nivo 35 监测每孔在340nm处的吸光度,连读1小时,每次读数间隔为1分钟。
[0120]
10)结果分析:不同浓度化合物对葡萄糖激酶激动活性的计算。
[0121]
: 阳性对照吸光度随检测时间变化的斜率的平均值。
[0122]
: 阴性对照吸光度随检测时间变化的斜率的平均值。
[0123]
: 化合物在相同浓度下吸光度随检测时间变化的斜率的平均值。
[0124][0125]
11)结果分析:ec
50
值的计算
[0126]
使用graphad 8.0,利用以下非线性拟合公式来得到化合物的ec50。 y=bottom (top
‑
bottom)/(1 10^((logec50
‑
x)*hillslope)) x:化合物浓度log值;y:化合物活性率。测试结果如下:
[0127]
表1.化合物gk激动活性测试结果
[0128]
化合物编号ec
50
dorzagliatin353化合物1406.2化合物2335化合物3463.8化合物4372化合物5363
[0129]
实施例7:体外肝微粒体代谢稳定性试验
[0130]
1)人肝微粒体(20mg蛋白/ml)和sd大鼠肝微粒体(20mg蛋白/ml) 采购于供应商corning。
[0131]
2)取化合物适量,先用dmso配制成5mm的储备液,再用50%甲醇
ꢀ‑
水稀释成100μm的测试化合物工作溶液,待用。
[0132]
3)从
‑
80℃冰箱中取出肝微粒体(20mg蛋白/ml),置于37℃水浴恒 温振荡器上预
温孵3min,融化待用。
[0133]
4)按照mgcl2
‑
pb溶液(6mm),肝微粒体(2.0mg蛋白/ml),0.8mg 蛋白/ml(咪达唑仑)的比例,制备温孵体系混合溶液(不含β
ꢀ‑
nadph)。
[0134]
5)制备100μm的测试化合物工作溶液,备用。
[0135]
6).对照组(不含β
‑
nadph):分别取25μl pb溶液到75μl(4) 所述温孵体系混合液中,涡旋30s,混匀,反应总体积100μl, 复样。放入到37℃水浴恒温振荡器中进行孵育,并开始计时, 取样时间点为0min和60min。
[0136]
7)样品组:分别取25μlβ
‑
nadph溶液(4mm)加入75μl(2) 所述反应体系中,涡旋30s,混匀,反应总体积100μl,复样。 放入到37℃水浴恒温振荡器中进行孵育,并开始计时,取样时 间点为0min,5min,15min,30min,60min。
[0137]
8)于各个时间点将样品管中取出,加入300μl冷的终止剂(含内 标),终止反应。
[0138]
9)涡旋5min后,离心10min(5500
×
g)。
[0139]
10)取上清液150μl加入150μl水,涡旋混匀,lc
‑
ms/ms进样 分析。
[0140]
11)数据分析
[0141]
用下列一级动力学公式计算半衰期(t1/2)和清除率(cl)
[0142]
c
t
=c0*e
‑
kt
[0143]
c
t
=(1/2)*c0[0144]
t
1/2
=ln2/k=0.693/k
[0145]
vd=1/肝微粒体中蛋白含量
[0146]
cl
int(liver)
=cl
int(mic)
×
肝重体重比
×
每克肝脏中的肝微粒体
[0147]
蛋白浓度
[0148]
测试结果如下:
[0149]
表2.化合物肝微粒体稳定性测试结果
[0150][0151][0152]
实施例8:体外肝细胞代谢稳定性试验
[0153]
1)人肝细胞(存活率92%)购于上海权阳,大鼠肝细胞(存活率90%) 购于瑞德肝脏疾病研究(上海)有限公司。
[0154]
2)称取适量取化合物,用dmso配制成5mm的储备液,再用50%甲 醇
‑
水稀释成100μm的测试化合物工作溶液,待用。
[0155]
3)从液氮生物容器中取出细胞,复苏细胞,采用台盼蓝染色法对 细胞进行计数,
然后将其稀释为活细胞数为2.0x10
6 cell/ml。
[0156]
4)制备2μm的测试化合物工作溶液,备用。
[0157]
5)预热给药工作液和肝细胞溶液。
[0158]
6)0分钟时间点试验方法:取25μl 2μm给药溶液,加入到样品 稀释板(96孔透明深孔板)相应孔中,再加入300μl含内标的 acn,然后加入25μl预热的肝细胞溶液,涡旋混匀,置于4℃ 冰箱保存。
[0159]
7)剩余时间点试验方法:分别将150μl给药工作溶液和150μl 预热的肝细胞溶液加入到已标记好的孵育管中,孵育体系活细 胞数为1.0x10
6 cell/ml,轻轻吹打,并放入二氧化碳培养箱 中的摇床上,转速200rpm,立即计时。分别于15分钟、30 分钟、60分钟、120分钟时,取50ul反应液加入含300μl的 acn(含格列本脲内标)的样品稀释板中相应位置,涡旋混匀。
[0160]
8)取上清液150μl加入150μl水,涡旋混匀,lc
‑
ms/ms进样 分析。
[0161]
9)数据分析
[0162]
用下列一级动力学公式计算半衰期(t1/2)和清除率(cl)
[0163]
c
t
=c0*e
‑
kt
[0164]
c
t
=(1/2)*c0[0165]
t
1/2
=ln2/k=0.693/k
[0166]
clint(hep)=ke/肝细胞量(million cells/ml)
[0167]
clint(liver)=clint(hep)
×
肝重体重比
×
每克肝脏中 的肝细胞数量
[0168]
测试结果如下:
[0169]
表3.化合物肝细胞稳定性测试结果
[0170][0171]
实施例9:大鼠体所内药代动力学试验
[0172]
<口服吸收性的研究实验材料和方法>
[0173]
1)使用动物使用sd大鼠。
[0174]
2)饲养条件使sd大鼠自由摄取固形饲料和纯净水。
[0175]
3)施与量、分组的设定利用规定的给药量进行口服给药、静脉给 药。如以下这定组。(各化合物的给药量有变化) 口服给药10mg/kg(n=5
‑
6)静脉给药1mg/kg(n=5
‑
6)
[0176]
4)给药溶液的制备口服给药为悬浮液、灌胃给药。静脉给药为溶液、 尾静脉给药。
[0177]
5)评价项目经时地采血,使用lc/ms/ms测定血浆中药物浓度。
[0178]
6)统计分析对于血浆中浓度变化,使用非线性最小二乘法程序算出 血浆中浓度时间曲线下面积(auc),由口服给药组和静脉给药组的 auc算出生物利用度(ba),和并统计静脉给药血药浓度消除半衰期 t
1/2
。
[0179]
测试结果如下:
[0180]
表4.化合物大鼠pk测试结果
[0181][0182]
对于本领域技术人员,本公开不只局限于前述说明性实施例,在 不脱离其必要属性的情况下能以其它特定形式体现。因此期望认为, 所有方面均作为说明性而不是限制性、对所附权利要求进行参考的实 施例而不是前述实施例,引用文献只是针对附加的权利要求而不是 上述的实例,以及落入权利要求等效性的含义和范围之内的所有变 化因此预期包含于此。
[0183]
本说明书中列举的所有专利、专利申请和文献参考均在此以其全 部内容引入作为参考。在不一致的情况下,包括定义的本公开将是有 说服力的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

