1.本发明涉及胰高血糖素样肽六连体的重组表达方法,尤其涉及提高胰高血糖素样肽六连体在毕赤酵母中表达水平或表达量的方法,属于胰高血糖素样肽六连体的重组表达领域。
背景技术:
2.糖尿病是以高血糖为特征的代谢性疾病,由胰岛素相对缺乏导致糖类代谢功能紊乱引起,肠源性激素胰高血糖素样肽
‑
1(glucagon
‑
like peptide
‑
1,glp
‑
1)因其依赖葡萄糖浓度促进胰岛β细胞分泌胰岛素来降低血糖水平,不会造成严重低血糖而引起广泛关注,但由于其在血液中的半衰期只有几分钟,限制了glp
‑
1在医疗上的应用。胰高血糖素样肽六连体(6
×
mglp
‑
1)是在已有的研究基础上,分别对天然glp
‑
1分子内dpp
‑
iv与trypsin敏感位点的氨基酸残基进行替换(his7‑
ala8突变为his7‑
ser8/gly8,lys
26,34
突变为gln
26
,asn
34
/asp
34
)、c末端添加cys、首尾相连形成6个串联重复、c末端添加his
‑
tag、密码子优化,并转化到毕赤酵母中,使其在毕赤酵母中分泌表达,但是其在毕赤酵母中表达水平低,使其生产和纯化的成本过高,不能满足工业生产的需求。
3.毕赤酵母是目前在工业生产重组蛋白中应用最为广泛一种酵母菌种,相对于其他表达系统,毕赤酵母表达系统具有可高密度发酵,操作简便快捷,同时作为真核生物又具备了原核生物所没有的特性,如蛋白的翻译后加工和修饰,包括二硫键形成、糖基化和脂酰化等。对于一些有细胞毒性的外源重组蛋白,可以将蛋白定位于毕赤酵母的过氧化物酶体中,既可以保证毕赤酵母不被毒性伤害,又可以防止蛋白酶对外源蛋白的降解,对于分泌到胞外的重组蛋白来说,由于毕赤酵母本身的分泌蛋白较少,在后期外源蛋白的分离和纯化可简化操作。极适合工业大规模发酵生产。但随着毕赤酵母表达系统的广泛应用发现,并不是所有外源蛋白基因均能在毕赤酵母中高效表达,传统方法如使用强启动子、密码子优化、增加拷贝数、基因的融合表达、优化发酵条件等对提高大多外源蛋白在毕赤酵母中的表达水平十分有限,甚至起不到作用。
4.由于6
×
mglp
‑
1在毕赤酵母中表达水平低,使其生产和纯化的成本过高,该缺陷严重限制了6
×
mglp
‑
1在医药上的广泛应用,亟待改进。
技术实现要素:
5.本发明的目的之一是提供提高胰高血糖素样肽六连体(6
×
mglp
‑
1)在毕赤酵母中表达水平的方法;本发明的目的之二是提供一种胰高血糖素样肽六连体基因酵母表达载体;本发明的目的之三是提供一种gese基因编辑载体。
6.本发明的目的之四是将胰高血糖素样肽六连体基因酵母表达载体以及gese基因编辑载体应用于提高胰高血糖素样肽六连体(6
×
mglp
‑
1)在毕赤酵母中表达水平。
7.本发明的上述目的是通过以下技术方案来实现的:
本发明一方面提供了一种提高胰高血糖素样肽六连体(6
×
mglp
‑
1)在毕赤酵母中表达水平的方法,包括:构建胰高血糖素样肽六连体酵母表达载体;将所构建的胰高血糖素样肽六连体酵母表达载体转化到宿主毕赤酵母菌株中诱导胰高血糖素样肽六连体在宿主毕赤酵母菌株中进行重组表达;其中,在诱导胰高血糖素样肽六连体在宿主毕赤酵母菌株中进行重组表达时,将宿主毕赤酵母菌株中的gese基因敲除或进行突变。
8.作为本发明一种优选的具体实施方案,本发明按照酵母偏好密码子,人工合成优化6
×
mglp
‑
1基因的dna序列,其核苷酸序列为seq id no.1所示;所述的gese基因的核苷酸序列为seq id no.1所示。
9.作为本发明一种优选的具体实施方案,所述的宿主毕赤酵母菌株优选为毕赤酵母表达菌株gs115。
10.作为本发明一种优选的具体实施方案,将宿主毕赤酵母菌株中的gese基因敲除或进行突变可采用crispr/cas9基因编辑技术,譬如,针对gese基因的靶序列构建gese基因编辑载体,将该gese基因编辑载体载体转化到含有胰高血糖素样肽六连体基因的宿主毕赤酵母表达菌株中,将宿主毕赤酵母表达菌株中的gese基因敲除;或者是构建gese基因敲除载体,将宿主毕赤酵母表达菌株中的gese基因敲除或进行突变。
11.作为一种参考的实施方式,本发明提供了胰高血糖素样肽六连体基因酵母表达载体的构建方法,包括:将胰高血糖素样肽六连体基因和α信号肽序列与ecori和noti双酶切线性化载体pgap9k(也可以是其他外源蛋白的真核表达载体为起始质粒)在同源重组酶作用下通过同源重组后转化大肠杆菌感受态,挑选阳性菌株得到胰高血糖素样肽六连体基因酵母表达载体,该载体命名为pgap9k
‑6×
mglp
‑
1,其结构图谱如图1所示,其中包括酵母组成型启动子gap,位于启动子下游的编码α
‑
因子分泌信号肽的核苷酸序列以及所述编码胰高血糖素样肽六连体的基因,所述编码胰高血糖素样肽六连体蛋白的基因5’端具有kex2基因表达产物切割识别序列aaaaga。pgap9k表达载体启动子后含有ecori和noti酶切位点可以将载体线性化,将人工合成的胰高血糖素样肽六连体基因以及α信号肽分别合成引物,其中α信号肽引物的5’端含有与载体ecori位点上游的25个重叠碱基,α信号肽3’端引物含有与6
×
mglp
‑
1重叠的25 bp碱基,外源基因6
×
mglp
‑
1的3’端引物含有与载体ecori位点上游的25个重叠碱基。
12.作为本发明一种优选的具体实施方案,将sali线性化后的pgap9k
‑6×
mglp
‑
1表达载体可通过电击转化导入毕赤酵母基因组中。
13.本发明还提供了一种采用crispr/cas9基因编辑方法针对gese基因的靶序列构建基因编辑载体的方法,包括:根据gese基因序列设计得到seq id no.3所示的靶位点序列,根据该靶位点序列设计引物进行pcr扩增:其中,所述的引物的序列为seq id no.4
‑ꢀ
seq id no.11所示;将pcr扩增产物与经酶切线性化的crispr/cas9质粒通过同源重组转化大肠杆菌,挑选阳性菌株,得到基因编辑载体,命名为bb3ch
‑
crispr载体。
14.作为本发明一种优选的具体实施方案,可以将bb3ch
‑
crispr载体进行电击转化到含有胰高血糖素样肽六连体基因的毕赤酵母表达菌株gs115中,获得gese基因敲除的胰高血糖素样肽六连体高表达菌株;其中,所述的电击转化条件优选为1500v,200ω,25μf;在ypd固体培养基(含有100μg/ml 潮霉素b, 50μg/ml cb),30℃培养3d;利用基因组pcr扩增
单菌落中的目的基因,经测序挑选靶序列位置具有测序重叠峰的菌株,挑取该菌株于ypd液体培养基(50μg/ml cb)中30℃ 200 rpm 培养至od600值1.5~2.0,将菌液按照千分之一接种量接种于新的ypd液体培养基(含有50μg/ml cb)中,重复三次;将最终得到的菌液于ypd固体培养基(含有50μg/ml cb)平板上划线,30℃培养3d;挑取单菌落进行基因组pcr,pcr产物测序,挑取gese基因序列编辑正确的菌种培养保存,得到基因组编辑后的6
×
mglp
‑1‑
gs115
‑
gese
‑
cri菌株。
15.本发明还提供了一种转基因酵母菌落pcr的分子生物学鉴定的过程,包括:得到转基因酵母单菌落之后,挑取菌体至20μl的0.02m naoh中100℃ 5分钟,取2μl上清作模板于20 μl pcr反应体系中检测目的基因编辑情况,采用western blot及elisa检测胰高血糖素样肽六连体在gs115中的表达水平是否提升。
16.本发明基于转录组测序(rna
‑
seq)分析低水平表达菌株(6
×
mglp
‑1‑
gs115)和高水平表达菌株(t4
‑
gs115)的差异基因,筛选一个转录水平差异显著的新基因gese。目前,还没有关于基因gese(pas_chr1
‑
3_0003)的研究报道,而且该基因在go以及ncbi数据库中没有对该基因的信息注释。本发明利用毕赤酵母crispr/cas9基因敲除技术敲除毕赤酵母宿主中gese基因后发现6
×
mglp
‑
1在毕赤酵母菌株gs115中的表达水平得到显著提高,该发现对于6
×
mglp
‑
1的产业化和扩大毕赤酵母的工业化应用范围具有重要价值,为提高外源蛋白基因在毕赤酵母中的表达水平提供基础研究和新途径。
附图说明
17.图 1 含有6
×
mglp
‑
1的酵母表达载体pgap9k
‑6×
mglp
‑
1的质粒图谱,6
×
mglp
‑
1基因以pgap启动子构建表达框。
18.图26
×
mglp
‑1‑
gs115基因组编辑所用crispr/cas9载体bb3ch
‑
crispr。
19.图 3crispr编辑前后基因序列比对。
20.图 4crispr编辑后测序峰图。
21.图 5western blot鉴定目的基因编辑前后6
×
mglp
‑
1表达, glp:6
×
mglp
‑
1基因在毕赤酵母宿主中的表达,其中,毕赤酵母宿主中的gese基因未进行敲除或突变;gese
‑
cri:6
×
mglp
‑
1基因在毕赤酵母宿主中的表达,其中,毕赤酵母宿主中的gese基因通过基因编辑被敲除。
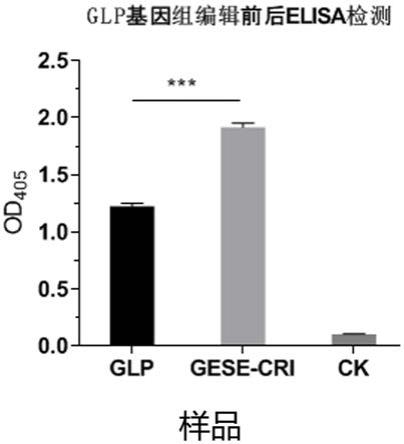
22.图 6elisa检测目的基因编辑后6
×
mglp
‑
1表达水平变化,ck: pgap9k空载体转化的gs115菌株作为阴性对照;glp:6
×
mglp
‑
1基因在毕赤酵母宿主中的表达水平,其中,毕赤酵母宿主中的gese基因未进行敲除或突变;gese
‑
cri:6
×
mglp
‑
1基因在毕赤酵母宿主中的表达水平,其中,毕赤酵母宿主中的gese基因通过基因编辑被敲除;elisa数据采用graphpad进行统计分析并作图,***代表p<0.01为极显著。
具体实施方式
[0023] 以下结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
[0024]
实施例1含有6
×
mglp
‑
1外源蛋白基因的表达载体的构建表达载体(pgap9k)启动子后含有ecori和noti酶切位点可以将载体线性化,将人工合成的6
×
mglp
‑
1以及α信号肽分别合成引物,其中α信号肽引物的5’端含有与载体ecori位点上游的25个重叠碱基,α信号肽3’端引物含有与6
×
mglp
‑
1重叠的25 bp碱基,外源基因6
×
mglp
‑
1的3’端引物含有与载体ecori位点上游的25个重叠碱基,高保真酶扩增外源基因6
×
mglp
‑
1和α信号肽序列,胶回收后与ecori和noti双酶切线性化载体50℃ 15 min孵育,在同源重组酶作用下通过同源重组后转化大肠杆菌dh5α感受态,挑选阳性菌株得到pgap9k
‑6×
mglp
‑
1载体,图1 为含有6
×
mglp
‑
1的酵母表达载体pgap9k
‑6×
mglp
‑
1的质粒图谱。
[0025]
实施例2含gese靶序列crispr/cas9载体bb3ch
‑
crispr的构建根据目的基因gese序列,在e
‑
crisp design网设计得到seq id no.3所示的靶位点序列,根据靶位点序列合成引物,引物序列见表1。
[0026]
表1 引物序列将扩增的产物与经bpii酶切线性化的crispr/cas9质粒通过同源重组转化大肠杆菌感受态dh5α,于lb固体培养基(含有50 μg/ml, 潮霉素b)中涂板,37℃倒置培养,菌落pcr挑选阳性菌株。图2为6
×
mglp
‑1‑
gs115基因组编辑crispr/cas9载体bb3ch
‑
crispr的图谱。
[0027]
实施例3电击法转化酵母菌株gs115(1)、将载体pgap9k
‑6×
mglp
‑
1使用sali酶切线性化,加入1/10v的3 m 醋酸钠和2.5v的无水乙醇混匀,12,000 rpm离心5 min,75%乙醇洗涤两次,晾干用10 μl ddh2o溶解
备用。
[0028]
(2)、挑取gs115单菌落于5ml液体ypd(含有50μg/mlcb)中30℃,200rpm培养a
600
值至1.4,5000rpm离心3min收集菌体,预冷的无菌水洗涤菌体两次,预冷的1m山梨醇溶液洗涤菌体一次,将菌体用100μl预冷1m山梨醇溶液重悬得到gs115感受态。取80μl的gs115感受态与10μl质粒轻弹管壁混合,加入2cm电击杯中,冰上10min,1500v,200ω,25μf电击,加入300μl预冷的1m山梨醇溶液混匀吸取至无菌2.0ml的ep管中,30℃静置1h,加入300μl液体ypd溶液,30℃,200rpm培养1h,200μl每板于ypd固体培养基(含有500μg/mlg418,50μg/mlcb)涂板,30℃倒置培养至长出单菌落。
[0029]
(3)、挑取单菌落菌体至20μl的0.02mol/l的naoh中重悬,100℃孵育5min,取2μl上清为模板于20μl的pcr体系中进行pcr鉴定,凝胶电泳,挑选pgap9k
‑6×
mglp
‑1‑
gs115阳性菌株。
[0030]
实施例4crisp载体bb3ch
‑
crispr电击法转化pgap9k
‑6×
mglp
‑1‑
gs115菌株及筛选(1)、接种bb3ch
‑
crispr大肠菌株于lb液体培养基(含50μg/ml潮霉素b)10ml,过夜培养,提取质粒,备用。
[0031]
(2)、菌株pgap9k
‑6×
mglp
‑1‑
gs115感受态的制备(制备方法同gs115感受态制备)。
[0032]
(3)、取bb3ch
‑
crispr质粒10μl(约1
‑
2μg,无需线性化),加入80μlpgap9k
‑6×
mglp
‑1‑
gs115感受态,轻弹管壁混匀,加入到2cm电击杯中,冰上10min,电击(电击条件同gs115转化),后续操作同转化gs115相同,100μl每板,于ypd固体培养基(含100μg/ml潮霉素b)涂板,30℃培养至长成单菌落。
[0033]
实施例5crispr/cas9基因编辑菌株的检测(1)、牙签挑取单菌落菌体于20μl0.02mol/l的naoh溶液中重悬,100℃5min,取2μl上清为模板于20μlpcr体系中,以目的基因靶序列上下游约100bp处合成的引物进行pcr检测,将pcr产物进行测序。
[0034]
(2)、测序结果与目的基因序列进行比对,靶序列处碱基改变且具有重叠峰的样本,挑取单菌落至ypd液体培养基(含50μg/mlcb)30℃,200rpm无筛选压培养至a
600
值1.5~2.0,千分之一接种至新的ypd液体培养基(含50μg/mlcb),培养至a
600
值1.5~2.0时,再次千分之一接种至新的ypd液体培养基(含50μg/mlcb),培养至a
600
值1.5~2.0,将菌液于ypd固体培养基(含50μg/mlcb)平板上划线,30℃培养至长成单菌落。
[0035]
(3)、菌落pcr,pcr产物测序,目的基因靶序列正确编辑的单菌落进行扩培并保菌。此时也可以提取酵母基因组,进行基因组pcr,进一步测序检测目的基因是否正确编辑。
[0036]
图5为elisa检测目的基因编辑前后6
×
mglp
‑
1表达水平变化结果,根据该试验结果可见,将毕赤酵母宿主中的gese基因通过基因编辑敲除后,6
×
mglp
‑
1基因在毕赤酵母宿主中的表达水平有显著提升。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。