1.本发明涉及有机合成技术领域,更具体地,涉及一种硫醚化芳杂环化合物及其制备方法和应用。
背景技术:
2.芳杂基化合物及其衍生物表现出有趣的生物活性,特别是杂芳基硫化物结构基序,在许多药物活性化合物和高级材料中均有发现。因此,在芳杂环中引入亚磺酰基具有重大的研究意义。但是通常c
‑
s构建最强有力的反应之一是硫醇或二硫化物与芳基卤化物或假卤化物的交叉偶联,该类反应往往需要钯、铜、钴的等过渡金属的催化(xu,x.,et al.(2013).“nickel
‑
mediated inter
‑
and intramolecular c
–
s coupling of thiols and thioacetates with aryl iodides at room temperature”organic letters15(3):550
–
553)。hostier等人报道了一种不使用有机金属或者过渡金属作为催化剂构造c
‑
s的方法,该方法是通过反应试剂的预官能化来实现的(hostier,t.,et al.(2015)."tfa
‑
promoted direct c
‑
h sulfenylation at the c
2 position of non
‑
protected indoles."chemical communications 51(73):13898
‑
13901.),该方法虽然避免了使用过渡金属作为催化剂,但是预官能化反应试剂的引入,增加了反应原料的获取的难度和成本,不利于工业生产。
技术实现要素:
3.本发明的首要目的是克服上述现有技术的不足,提供一种硫醚化芳杂环化合物的制备方法,该方法无需使用过渡金属作为催化剂,也无需预官能化的反应试剂,具有节能环保、原料易得的优势。
4.本发明的另一目的是提供一种硫醚化芳杂环化合物。
5.本发明的进一步目的是提供所述硫醚化芳杂环化合物的应用。
6.本发明的上述目的通过以下技术方案实现:
7.一种硫醚化芳杂环化合物的制备方法,包括如下步骤:
8.将芳杂环化合物(i)、硫酚(ii)以及氧化剂溶于有机溶剂中,在反应温度为0~100℃的条件下反应制得所述硫醚化芳杂环化合物;所述芳杂环化合物(i)和硫酚(ii)的分子结构式如下:
[0009][0010]
其中,r1选自烷基、烷氧基、卤素、芳基、氰基、硝基或羟基;r2选自烷基、烷氧基、卤素、芳基、硝基或羟基;
[0011]
所述氧化剂选自selectfluor氟试剂,[双(三氟乙酰氧基)碘]苯(pifa),碘苯二乙
酸酯(pida)中的一种或多种。
[0012]
优选地,所述芳杂环化合物(i)的分子结构式如下:
[0013][0014]
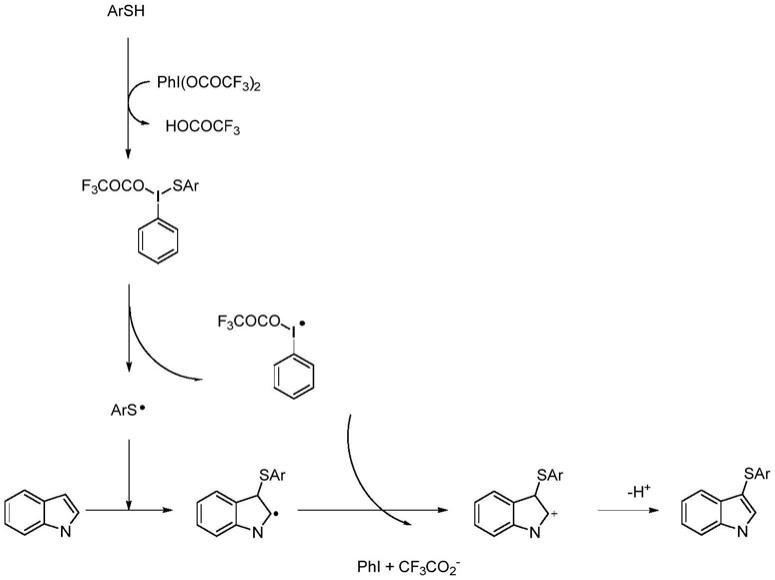
下面以pifa作为氧化剂的方案来说明硫醚化芳杂环化合物的合成机理:
[0015][0016]
本发明中,硫酚在pifa作用下产生了硫自由基和碘自由基,硫自由基进攻吲哚的c3形成自由基中间体。中间体再被前步生产碘自由基氧化形成阳离子中间体,最后去质子化形成目标产物。
[0017]
本发明所述硫醚化芳杂环化合物的制备方法在0~100℃条件下反应结束后还包括后处理步骤,所述后处理步骤依次为洗涤、萃取、柱层析。
[0018]
优选地,所述芳杂环化合物(i)与硫酚(ii)的摩尔比为1:(1~5)。更优选为1:1。
[0019]
优选地,所述芳杂环化合物(i)与氧化剂的摩尔比为1:(1~5)。
[0020]
优选地,所述反应温度为20~60℃,反应时间为6~24h。更优选地,反应温度为30℃。经过多次实验,发明人发现反应温度为20~60℃时,产物产率较高,最佳反应温度为30℃。
[0021]
优选地,所述有机溶剂选自二氯甲烷、乙腈或1,4
‑
二氧六环中的一种或多种。更优选为二氯甲烷。
[0022]
优选地,所述氧化剂选自[双(三氟乙酰氧基)碘]苯。
[0023]
一种硫醚化芳杂环化合物,由上述方法制备得到。
[0024]
本发明还保护上述硫醚化芳杂环化合物在合成抗肿瘤药物、抗癌药物、有机光电功能材料中间体领域中的应用。
[0025]
与现有技术相比,本发明的有益效果是:
[0026]
本发明提供了一种硫醚化芳杂环化合物的制备方法,所述方法无需使用过渡金属作为催化剂,也无需使用预官能化反应试剂,可在较温和的条件下进行,具有绿色环保、成本低、底物选择性广、易于工业化的优势,是一种环境友好型的方法。
附图说明
[0027]
图1是合成硫醚化的芳杂环化合物的分子结构式。
[0028]
图2是实施例1~7制得的3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的核磁h谱。
[0029]
图3是实施例1~7制得的3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的核磁c谱。
[0030]
图4是实施例8制得的n
‑
甲基
‑3‑
(对甲苯甲硫基)
‑
1h
‑
吲哚的核磁h谱。
[0031]
图5是实施例8制得的n
‑
甲基
‑3‑
(对甲苯甲硫基)
‑
1h
‑
吲哚的核磁c谱。
[0032]
图6是实施例9制得的2
‑
苯基
‑5‑
(对甲苯甲硫基)噻吩的核磁h谱。
[0033]
图7是实施例9制得的2
‑
苯基
‑5‑
(对甲苯甲硫基)噻吩的核磁c谱。
具体实施方式
[0034]
为了更清楚、完整的描述本发明的技术方案,以下通过具体实施例进一步详细说明本发明,应当理解,此处所描述的具体实施例仅用于解释本发明,并不用于限定本发明,可以在本发明权利限定的范围内进行各种改变。
[0035]
实施例1
[0036]
一种硫醚化芳杂环化合物,其分子结构式如图1中式(a)所示。
[0037]
上述硫醚化芳杂环化合物的制备方法,包括如下步骤:
[0038]
取15ml耐压反应管,加入pifa 90mg,吲哚24mg,4
‑
甲基苯硫酚30mg,二氯甲烷2ml,敞口反应,于30℃下搅拌过夜。反应结束后加入乙酸乙酯10ml淬灭反应,加盐水10ml洗涤,分出有机相,水相用乙酸乙酯萃取3次,合并有机相,柱层析分离得到3
‑
(对甲苯硫基)
‑
1h
‑
吲哚纯品38.4mg,产率80%。
[0039]
实施例2
[0040]
与实施例1不同的是,本实施例采用乙腈代替二氯甲烷。本实施例中3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的产率为50%。
[0041]
实施例3
[0042]
与实施例1不同的是,本实施例采用selectfluor氟试剂代替pifa。本实施例中3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的产率为45%。
[0043]
实施例4
[0044]
与实施例1不同的是,本实施例采用碘苯二乙酸酯(pida)代替pifa。本实施例中3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的产率为53%。
[0045]
实施例5
[0046]
与实施例1不同的是,本实施例采用1,4
‑
二氧六环代替二氯甲烷。本实施例中3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的产率为30%。
[0047]
实施例6
[0048]
与实施例1不同的是,本实施例温度为20℃代替30℃。本实施例中3
‑
(对甲苯硫
基)
‑
1h
‑
吲哚的产率为70%。
[0049]
实施例7
[0050]
与实施例1不同的是,本实施例温度为60℃代替30℃。本实施例中3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的产率为68%。
[0051]
实施例8
[0052]
一种硫醚化芳杂环化合物,其分子结构式如图1中式(b)所示。
[0053]
上述硫醚化芳杂环化合物的制备方法,包括如下步骤:
[0054]
取15ml耐压反应管,加入pifa90mg,n
‑
甲基吲哚27mg,4
‑
甲基苯硫酚30mg,二氯甲烷2ml,敞口反应,于30℃下搅拌过夜。反应结束后加入乙酸乙酯10ml淬灭反应,加盐水10ml洗涤,分出有机相,水相用乙酸乙酯萃取3次,合并有机相,柱层析分离得到n
‑
甲基
‑3‑
(对甲苯甲硫基)
‑
1h
‑
吲哚纯品35mg,产率70%。
[0055]
实施例9
[0056]
一种硫醚化芳杂环化合物,其分子结构式如图1中式(c)所示。
[0057]
上述硫醚化芳杂环化合物的制备方法,包括如下步骤:
[0058]
取15ml耐压反应管,加入pifa 90mg,邻苯噻吩32mg,4
‑
甲基苯硫酚30mg,二氯甲烷2ml,敞口反应,于30℃下搅拌过夜。反应结束后加入乙酸乙酯10ml淬灭反应,加盐水10ml洗涤,分出有机相,水相用乙酸乙酯萃取3次,合并有机相,柱层析分离得到2
‑
苯基
‑5‑
(对甲苯甲硫基)噻吩纯品48mg,产率58%。
[0059]
对比例1
[0060]
一种硫醚化芳杂环化合物的制备方法,包括如下步骤:
[0061]
取15ml耐压反应管,加入n
‑
甲基吲哚27mg,n
‑
(硫酚)丁二酰亚胺40mg,三氟乙酸342mg,二氯甲烷2ml,闭口反应,于室温下搅拌4
‑
6。反应结束后加入乙酸乙酯10ml淬灭反应,加盐水10ml洗涤,分出有机相,水相用乙酸乙酯萃取3次,合并有机相,柱层析分离得到n
‑
甲基
‑2‑
(硫酚)
‑
1h
‑
吲哚纯品35mg,产率81%。
[0062]
本对比例提供了一种硫醚化芳杂环化合物的制备方法,该方法以预官能化试剂n
‑
(硫酚)丁二酰亚胺作为原料,存在原料获取难、成本高以及不利于工业化生产的问题。
[0063]
对比例2
[0064]
本对比例提供一种硫醚化芳杂环化合物的制备方法,与对比例1的区别在于,本对比例用硫酚(ii)代替n
‑
(硫酚)丁二酰亚胺。
[0065]
本对比例无法产生硫自由基,进而无法合成硫醚化芳杂环化合物。
[0066]
测试表征
[0067]
图2和图3分别是实施例1~5制得的3
‑
(对甲苯硫基)
‑
1h
‑
吲哚的核磁h谱和核磁c谱。图2氢谱峰的化学位移1h nmr(400mhz,cdcl3)δ8.37(s,1h),7.63(d,j=7.9hz,1h),7.46(d,j=2.6hz,1h),7.43(d,j=8.1hz,1h),7.30
–
7.26(m,1h),7.19
–
7.15(m,1h),7.07
–
7.03(m,2h),6.99(d,j=8.2hz,2h),2.26(s,3h);图3碳谱中谱峰的化学位移
13
c nmr(100mhz,cdcl3)δ136.5,135.5,134.7,129.1,126.3,123.0,120.9,119.7,111.6,103.5,20.9.证实所得物质为3
‑
(对甲苯硫基)
‑
1h
‑
吲哚。
[0068]
图4和图5分别是实施例8制得的n
‑
甲基
‑3‑
(对甲苯甲硫基)
‑
1h
‑
吲哚的核磁h谱和核磁c谱。从图4中可知,1h nmr(400mhz,cdcl3)δ7.60(d,j=7.9hz,1h),7.32(d,j=8.2hz,
1h),7.29
–
7.24(m,1h),7.18
–
7.14(m,1h),7.03(d,j=8.3hz,2h),6.97(d,j=8.3hz,2h),3.81(s,3h),2.24(s,3h);从图5中可知,
13
c nmr(100mhz,cdcl3):δ136.1,135.0,134.3,132.5,126.7,122.7,121.3,119.3,109.5,100.0,30.7,20.9。证实所得物质为n
‑
甲基
‑3‑
(对甲苯甲硫基)
‑
1h
‑
吲哚。
[0069]
图6和图7分别是实施例9制得的2
‑
苯基
‑5‑
(对甲苯甲硫基)噻吩的核磁h谱和核磁c谱。从图6可知,1h nmr(400mhz,cdcl3)δ7.61
–
7.53(m,2h),7.38(dd,j=8.1,6.8hz,2h),7.31(d,j=7.3hz,1h),7.26
–
7.21(m,4h),7.10(d,j=8.0hz,2h),2.32(s,3h);从图7可知,
13
c nmr(100mhz,cdcl3)δ149.3,136.5,136.2,134.6,133.9,131.9,129.9,129.0,128.0,125.8,123.5,21.0。证实所得物质为2
‑
苯基
‑5‑
(对甲苯甲硫基)噻吩。
[0070]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。