1.本发明属于分子检测及高通量测序技术领域,涉及一种构建用于同源重组修复缺陷检测的靶标集合的方法。
背景技术:
2.卵巢癌是女性生殖系统常见的恶性肿瘤,其传统的标准疗法为肿瘤细胞减灭术联合以铂类为基础的化疗,尽管多数患者可经标准疗法获得临床缓解,但卵巢癌患者三年内复发率较高。
3.近年来,多聚苷二磷酸核糖聚合酶(parp)抑制剂的出现改变了卵巢癌患者的治疗格局,目前已有多款parp抑制剂上市,可显著延长卵巢癌患者的无进展生存时间,parp基因家族包括17个成员,其中parp1和parp2主要通过碱基切除修复途径在dna单链断裂修复中发挥重要作用。
4.brca1/2基因是dna损伤修复通路中的重要基因,该基因突变可抑制细胞dna损伤后的修复能力,引起同源重组缺陷(homologous recombination deficiency,hrd),brca功能缺失或其他同源重组通路基因发生突变或功能缺失,同源重组修复(homologous recombinant repair,hrr)出现缺陷,从而不能修复双链断裂的dna,最终导致癌变,在hrd肿瘤细胞中dna双链无法修复,parp抑制剂又阻断单链修复,从而形成“合成致死”效应,导致肿瘤细胞死亡,达到抗肿瘤的目的。
5.由于parp抑制剂的合成致死原理,临床上parp抑制剂的应用依赖于相应的生物标志物状态,目前最常见的parp抑制剂应用生物标志物包括brca基因突变、其他hrr基因突变和hrd,hrd可通过同源重组修复相关基因突变检测和基因组不稳定性两种方式判断,对于基因组不稳定的评估,目前可通过对三类不稳定事件的检测进行评估,包括杂合性缺失、大片段迁移及端粒等位基因不平衡。
6.对于包括brca1/2基因在内的hrr基因突变检测目前技术原理均较为简单,通过多重扩增子测序或靶向捕获测序均可实现,如cn110863048a公开了一种检测dna同源重组修复通路有效性的探针库、检测方法和试剂盒,使用该探针库对待分析的目标dna片段进行富集,然后再对富集后的dna片段通过测序手段进行测序,从而实现对基因检测的有效性,与dna同源重组修复通路相关的基因可用于预测肿瘤发生的倾向性、评估肿瘤恶性程度和临床预后、指导新药筛选与研发。
7.但brca1/2基因突变的卵巢癌患者仅占约20%
‑
30%,而在此基础上增加基因组不稳定性的评估,则可将同源重组缺陷阳性患者的比例提高至50%,将大大增加parp抑制剂的潜在获益人群,但对于基因组不稳定性的评估还存在较大的困难,这主要是由于基因组不稳定性需要对整个基因组进行监测,分析判断出哪些区域发生了杂合性缺失或拷贝数扩增等不稳定性事件。目前有一些采用全基因组测序(wgs)来进行相应的检测分析,但人类基因组约有3亿个碱基,仅低深度的测序也需要较大的测序数据量,将导致检测成本较高,经济成本效益极低。
8.综上所述,提供一种能够同时对hrr基因突变情况进行检测,同时又能以较低成本检测基因组不稳定性状态的产品,在最大化肿瘤患者parp抑制剂用药机会的同时,降低患者的检测经济负担。
技术实现要素:
9.针对现有技术的不足和实际需求,本发明提供一种构建用于同源重组修复缺陷检测的靶标集合的方法,本发明筛选能够充分表征基因组不稳定性的snp位点并结合同源重组修复(hrr)通路基因构建靶标集合,并设计探针组合物,使用所述探针组合物对待分析的目标dna片段进行富集,然后再对富集后的dna片段进行测序及数据分析,既能够进行hrr基因突变情况检测又能以较低成本检测基因组不稳定性,实现了全面评估同源重组修复缺陷。
10.为达上述目的,本发明采用以下方案:第一方面,本发明提供一种构建用于同源重组修复缺陷检测的靶标集合的方法,所述构建用于同源重组修复缺陷检测的靶标集合的方法包括以下步骤:(1)从chinamap中国万人基因组数据库的数据中筛选snp位点,筛选条件包括:位于非外显子区域、突变等位基因频率介于30%~70%、位点附近100 bp区域gc含量介于30%~70%且为非插入缺失变异的单位点突变;(2)从步骤(1)筛选到的snp位点中过滤掉位于基因组重复区和连锁不平衡位点,选择间隔距离均一的位点,所述间隔距离为50~300 kb,得到snp位点,所述snp位点包括10913个snp位点,各snp位点的id如表1所示;(3)将步骤(2)得到的snp位点与同源重组修复通路基因包括:arid1a、atm、atr、atrx、bap1、bard1、blm、brca1、brca2、brip1、cdk12、chek1、chek2、emsy、fam175a、fanca、fancc、fancd2、fance、fancf、fanci、fancl、fancm、mre11a、nbn、palb2、ppp2r2a、pten、rad50、rad51、rad51b、rad51c、rad51d、rad54b、rad54l、rpa1、slfn11、slx4、wrn和tp53bp1组合,得到所述用于同源重组修复缺陷检测的靶标集合。
11.表1
通过对snp位点进行探针捕获,根据位点的覆盖度比较情况判断是否发生了杂合性缺失、大片段重排或端粒等位基因不平衡,通过对这三种事件进行分析,将其加和作为代表基因组不稳定性的评分,从而分析基因组不稳定性。本发明针对10913个snp位点进行捕获,相较于wgs捕获区域大大减小,可显著降低所需测序数据量及成本,并且与wgs检测结果有较高的一致性,可应用于hrd检测。
12.本发明筛选能够充分表征基因组不稳定性的snp位点并结合同源重组修复(hrr)通路基因构建靶标集合,并设计探针组合物,利用所述探针组合物能够对hrr基因的变异进行检测,同时对基因组不稳定性进行准确评估,能够有效扩大同源重组修复缺陷阳性的检出率。
13.第二方面,本发明提供一种检测同源重组修复基因变异和基因组不稳定性的探针组合物,所述探针组合物包括第一方面中所述的同源重组修复通路基因的子探针组a(hrr基因子探针组)及snp位点的子探针组b(snp位点子探针组)。
14.所述子探针组a为针对第一方面所述的40个同源重组修复通路基因设计长度为120 bp的探针、并以头尾相接的形式覆盖基因的所有编码区,且探针靶向的区域还包括brca1和brca2基因的非翻译区(utr)。
15.所述子探针组a的具体设计方法如下:1)依据ucsc数据库获得40个基因的编码区的基因组对应序列,同时依据该数据库标记出其中的重复区域。所述重复区域包括sine,line及ltr等,对于这些重复区域,不进行探针设计;2)对于长度小于120 bp的基因编码区段,设计单条探针完全覆盖该编码区段,在此前提下可进行适当左右挪动,以使探针的gc含量在35%~65%;3)对于长度大于120 bp的基因编码区段,首先在区段的首尾各外延30 bp,获得新的目标区段n。对每个目标区段n,从第1 bp开始作为第一条探针的起点,探针与探针之间首尾相接,最终平铺覆盖整个目标区段n;4)将步骤3)中的每条探针进行blast与全基因组进行比对,对于存在5条以上超过90%同源的序列标记为风险探针,回溯该探针对应的基因组位置,左右挪动探针的位置,直至blast结果满足非风险探针的要求。
16.所述子探针组b为针对第一方面所述的10913个snp位点分别设计长度为120 bp的探针,针对每个位点设计一条探针,对应snp位点尽量接近探针的中间位置,并且探针序列的gc含量在40%~65%之间,避开基因组重复区域。
17.所述子探针组b的具体设计方法如下:1)依据ucsc数据库获得10913个snp位点的基因组坐标,以每个snp位点为中心,左右各60 bp的序列记为针对该位点的候选探针序列1;2)对候选探针序列1进行gc含量分析,若该探针的gc含量在40%~65%之间,则进行下一步分析;若gc含量低于40%或高于65%,则应左右挪动探针的位置,直至gc含量达到最佳要求,同时应保证snp位点在探针的覆盖范围内,所得序列为该位点的探针序列2;3)对探针序列2进行blast与全基因组进行比对,对于存在5条以上超过90%同源的序列标记为风险探针,回溯该探针对应的基因组位置,左右挪动探针的位置,直至blast结果满足非风险探针的要求。
18.以上方法设计出的探针,交由专业的探针合成公司进行合成,所有的探针5’端进行生物素修饰。
19.本发明中,所述检测同源重组修复基因变异和基因组不稳定性的探针组合物能够在较低测序量条件下,经济可靠地同时检出同源重组修复基因变异及基因组不稳定性。
20.优选地,所述子探针组a和子探针组b的探针摩尔浓度比为(2~5):1,包括但不限于2:1、3:1、4:1或5:1,优选为4:1。
21.本发明中,通过控制hrr基因子探针组与snp位点子探针组的混合比例,在有限的测序量条件下,使得hrr基因区与snp区获得不同的覆盖深度,又同时满足基因变异及不稳定性评估的深度要求,从而精确地判断同源重组缺陷状态,降低了检测成本,提高了检测经济效益。
22.第三方面,本发明提供第二方面述的检测同源重组修复基因变异和基因组不稳定性的探针组合物在制备同源重组修复缺陷检测产品中的应用。
23.第四方面,本发明提供一种同源重组修复缺陷检测试剂盒,所述同源重组修复缺陷检测试剂盒包括第三方面所述的检测同源重组修复基因变异和基因组不稳定性的探针组合物。
24.优选地,所述同源重组修复缺陷检测试剂盒还包括dna文库构建试剂、udi接头、杂交与洗脱试剂或杂交产物扩增试剂中的任意一种或至少两种的组合。
25.优选地,所述dna文库构建试剂包括dna片段化酶、连接酶或扩增引物中的任意一种或至少两种的组合。
26.优选地,所述udi接头包括udi adapter 1
‑
12。
27.优选地,所述杂交与洗脱试剂包括杂交缓冲液和/或阻断剂。
28.优选地,所述杂交产物扩增试剂包括pcr master mix和/或pcr引物。
29.优选地,所述同源重组修复缺陷检测试剂盒还包括核酸纯化试剂。
30.优选地,所述核酸纯化试剂包括磁珠和/或清洗液。
31.优选地,所述磁珠为链霉亲和素磁珠。
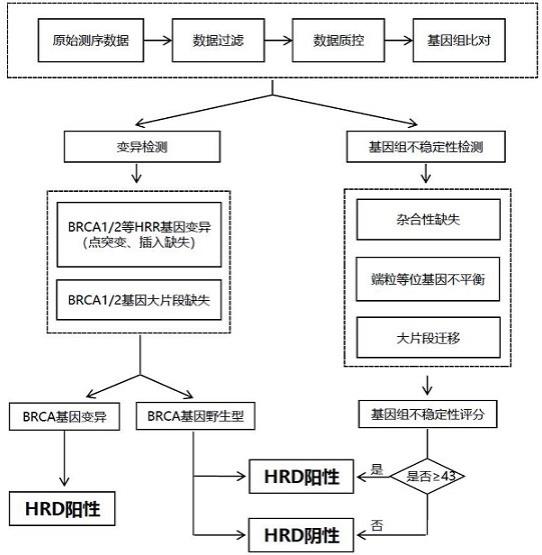
32.优选地,所述同源重组修复缺陷检测试剂盒的使用方法(流程如图2所示)包括以下步骤:(1)将样本dna片段化并进行末端修复补平加a,随后进行udi接头连接,纯化产物后进行pcr扩增,构建预文库;(2)利用第三方面所述的检测同源重组修复基因变异和基因组不稳定性的探针组合物与所述预文库进行杂交,得到靶标dna片段;(3)以所述靶标dna片段为模板利用pcr通用引物进行扩增,得到靶向捕获文库;(4)对所述靶向捕获文库进行双端测序,并对测序数据进行分析。
33.优选地,步骤(1)所述纯化包括磁珠纯化。
34.优选地,步骤(4)所述分析包括测序数据质控评价、基因点突变分析、插入/缺失变异注释、大片段重排和基因组杂合性缺失分析、大片段迁移分析和端粒等位基因不平衡分析。
35.优选地,hrr基因点突变及插入/缺失变异采用varscan软件基于启发式算法和统计学来进行分析,寻找变异位点。
36.优选地,所述基因组杂合性缺失分析、大片段迁移和端粒等位基因不平衡分析采用以下流程:1)对bam文件进行每个杂合子snp上的每个等位基因的等位型计数;2)统计snp探针区域的测序深度;3)采用r软件对每个snp的baf(b allele frequency,b等位基因频率)进行纠正校准,将校准后的baf和logr(log r ratio,对数比值)使用ascat进行基因组拷贝数变化和区域划分;4)将结果输入sequenza软件进行不同区域的基因组不稳定类型评估并分别统计基因组杂合性缺失、大片段迁移和端粒等位基因不平衡的数目。
37.优选地,同源重组修复缺陷阳性判断标准为具有brca1或brca2基因致病性突变或疑似致病突变和/或基因组不稳定性评分≥43,所述基因组不稳定性评分为基因组杂合性缺失、大片段迁移及端粒等位基因不平衡事件的加和。
38.所述同源重组修复缺陷(hrd)分析流程如图3所示。
39.与现有技术相比,本发明具有以下有益效果:(1)本发明筛选能够充分表征基因组不稳定性的snp位点并结合同源重组修复(hrr)通路基因构建靶标集合,并设计探针组合物,利用所述探针组合物能够对hrr基因的变异进行检测,同时对基因组不稳定性进行准确评估,能够有效扩大同源重组缺陷阳性的检出率,并降低检测成本;(2)本发明通过控制hrr基因子探针组和snp位点子探针组的混合比例,在有限的测序量条件下,使得hrr基因区与snp区获得不同程度的高深度覆盖,同时满足基因变异及不稳定性评估的深度要求,从而精确地判断患者同源重组缺陷状态,降低了检测成本,提高了检测经济效益。
附图说明
40.图1为本发明snp位点筛选流程图;图2为本发明检测流程图;图3为本发明hrd分析流程图;图4为hrr基因区和snp位点区平均测序深度结果图。
具体实施方式
41.为进一步阐述本发明所采取的技术手段及其效果,以下结合实施例和附图对本发明作进一步地说明。可以理解的是,此处所描述的具体实施方式仅仅用于解释本发明,而非对本发明的限定。
42.实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。
43.实施例1本实施例从来自临床合作单位的卵巢癌石蜡包埋(ffpe)肿瘤组织切片样本中提取dna,包括以下步骤:(1)脱蜡1)向蜡卷组织中加入1 ml二甲苯,盖上管盖,6档涡旋混匀10秒,50℃孵育5 min,室温下最大转速离心2 min,用枪头小心地吸掉上清;
2)向沉淀中加入1 ml无水乙醇,涡旋混匀,室温下最大转速离心2 min,用枪头小心的吸掉上清;3)置于56℃开盖放置,直到乙醇完全挥发;(2)裂解1)向步骤(1)产物中加入180 μl缓冲液atl重悬沉淀,加入20 μl蛋白酶k,涡旋混匀,56℃孵育1 h;2)振荡混匀,90℃放置1 h(需等温度升至90℃,再将样品放入孵育);3)将样品取出,室温放置2 min,恢复室温后瞬时离心;(3)清洗1)向步骤(2)产物中加入200 μl缓冲液al和200 μl无水乙醇,振荡混匀,最大转速离心2 min;2)将全部裂解产物转移至qiaamp minelute柱中(放在2 ml收集管里),不要弄湿边缘,盖上管盖,室温下最大转速离心1 min,弃掉滤液;3)将离心后的qiaamp minelute柱放置在新的2 ml收集管里,小心的打开qiaamp minelute柱的盖子,加入500 μl缓冲液aw1,不要弄湿边缘,盖上管盖,以最大转速离心1min,弃掉滤液;4)将离心后的qiaamp minelute柱放置在新的2 ml收集管里,小心的打开qiaamp minelute柱的盖子加入500 μl缓冲液aw2,不要弄湿边缘,盖上管盖,以最大转速离心1 min,弃掉滤液;5)将qiaamp minelute柱放在一个新的2 ml收集管里,最大转速离心3 min,使膜完全甩干;(4)收集将qiaamp minelute柱转移到新的1.5ml离心管中,开盖晾干4 min,去除吸附柱胶圈中的残液,向吸附柱中加入60 μl缓冲液eb,室温静置5 min后,最大转速离心1 min,洗脱液即为所提取dna。
44.实施例2本实施例采用实施例1核酸提取得到的dna,片段化后,进行接头连接、两轮纯化、pcr扩增和质控检测,构建预文库,步骤如下:(1)酶促反应将5 μl fx buffer和10 μl fx enzyme mix预混合,保存在冰上,与35 μl样本dna混合成50 μl总体系,将pcr仪预冷至4℃,并设置程序:32℃、32 min,65℃、30 min,4℃ hold,热盖70℃,pcr反应结束后将产物转移至冰上;(2)接头连接将15 μl水、20 μl缓冲液和10 μl连接酶进行预混合,并加入5 μl接头原液,与上步产物配制成100 μl总体系,在pcr仪中20℃孵育15 min,热盖关闭;(3)连接后纯化1)提前将ampure xp beads从4℃冰箱取出,置于室温平衡30 min,并振荡混匀使其充分重悬,向接头连接产物中加入0.8
×
的磁珠,涡旋混匀后室温下孵育5 min,上磁力架澄清后弃上清,加入200 μl 80%乙醇,1 min后弃上清,重复清洗一次;
2)在室温下将磁珠干燥4 min,晾干后用52.5 μl水重悬磁珠,室温下孵育5 min,上磁力架至液体澄清,转移50 μl上清液,向50 μl上清液中加入1
×
的磁珠,涡旋混匀后室温下孵育5 min;3)上磁力架澄清后弃上清,加入200 μl 80%乙醇,1 min后弃上清,重复清洗一次;4)在室温下将磁珠干燥4 min,晾干后用26 μl水重悬磁珠,室温下孵育5 min,上磁力架至液体澄清,转移23.5 μl上清液;(4)文库扩增1)向纯化后的接头连接产物中加入2.4 μl pcr primer mix,40 μl hifi pcr master mix和14.1 μl水的混合物,配制成总体积80 μl的pcr反应体系,pcr仪设置为98℃预变性2 min,98℃变性20 sec,60℃退火30 sec,72℃延伸30sec,扩增8 cycles,72℃完全延伸1 min,4℃ hold,热盖105℃;2)向扩增后产物中加入1
×
的磁珠,枪头吹打或涡旋混匀后室温下孵育5 min;3)上磁力架澄清后弃上清,加入200 μl 80%乙醇,1 min后弃上清,重复清洗一次;4)在室温下将磁珠干燥4 min,晾干后用30 μl水重悬磁珠,室温下孵育5 min,上磁力架至液体澄清,转移28 μl上清液,所得纯化后产物即为预文库。
45.实施例3本实施例采用实施例2所构建的预文库进行目的片段捕获,步骤如下:(1)向pooling后的预文库中加入8μl blockers、5μl blocker solution和5μl本发明的探针组合物(hrr基因子探针组a与snp位点子探针组b摩尔浓度比为4:1),置于真空浓缩仪中45℃蒸干,在此期间可将链霉亲和素磁珠和纯化磁珠取出,室温平衡30 min,其中,所述子探针组a的具体设计方法如下:1)依据ucsc数据库获得40个基因的编码区的基因组对应序列,同时依据该数据库标记出其中的重复区域。所述重复区域包括sine,line及ltr等,对于这些重复区域,不进行探针设计;2)对于长度小于120 bp的基因编码区段,设计单条探针完全覆盖该编码区段,在此前提下可进行适当左右挪动,以使探针的gc含量在35%~65%;3)对于长度大于120 bp的基因编码区段,首先在区段的首尾各外延30 bp,获得新的目标区段n。对每个目标区段n,从第1 bp开始作为第一条探针的起点,探针与探针之间首尾相接,最终平铺覆盖整个目标区段n;4)将步骤3)中的每条探针进行blast与全基因组进行比对,对于存在5条以上超过90%同源的序列标记为风险探针,回溯该探针对应的基因组位置,左右挪动探针的位置,直至blast结果满足非风险探针的要求;所述子探针组b的具体设计方法如下:1)依据ucsc数据库获得10913个snp位点的基因组坐标,以每个snp位点为中心,左右各60 bp的序列记为针对该位点的候选探针序列1;2)对候选探针序列1进行gc含量分析,若该探针的gc含量在40%~65%之间,则进行下一步分析;若gc含量低于40%或高于65%,则应左右挪动探针的位置,直至gc含量达到最佳要求,同时应保证snp位点在探针的覆盖范围内,所得序列为该位点的探针序列2;3)对探针序列2进行blast与全基因组进行比对,对于存在5条以上超过90%同源的
序列标记为风险探针,回溯该探针对应的基因组位置,左右挪动探针的位置,直至blast结果满足非风险探针的要求;以上方法设计出的探针,交由探针合成公司integrated dna technologies(idt)进行合成,所有的探针5’端进行生物素修饰;(2)将fast hybridization mix放置在65℃金属浴中孵育10 min至沉淀溶解,迅速涡旋后取20 μl加入蒸干的样本中,指尖轻弹混匀,室温静置5 min;(3)将上步反应快速离心去除气泡后,加入30 5μl hybridization enhancer至表面进行油封,放入pcr仪杂交,pcr仪程序设置为95℃ hold,95℃、5 min,60℃、4 h,60℃ hold;(4)将fast binding buffer,fast wash buffer1,fast wash buffer2放置48℃恒温金属浴上加热至溶解;(5)试剂预热:按照实际要求量分装fast wash buffer1, fast wash buffer2,并将其分别置于68℃和48℃中预热;(6)充分混匀链霉亲和素磁珠,取100 μl至1.5 ml离心管中,加入200 μl fast binding buffer,用枪头吹打混匀,置于磁力架至澄清,弃上清;重复加入并弃上清2次,最后加入200 μl fast binding buffer重悬链霉亲和素磁珠;(7)杂交4 h结束后,将清洗后的链霉亲和素磁珠拿到杂交管旁,迅速将50 μl杂交液转移至链霉亲和素磁珠中,振荡混匀后置于振荡器上室温充分混匀30 min;(8)向上步混合物中加入200 μl预热的fast wash buffer1,点震混匀,68℃孵育5 min,置于磁力架至液体澄清,弃上清,重复1次,将液体转移至新的1.5 ml离心管中,置于磁力架至液体澄清,弃上清;(9)向离心管中加入200 μl预热的fast wash buffer2,点震混匀,48℃孵育5min,置于磁力架至液体澄清,弃上清;重复2次,并用小枪将残液吸取干净,加入45 μl水,混匀备用;(10)取22.5 μl结合于链霉亲和素磁珠的杂交产物,并加入27.5 μl的扩增试剂混合物,配制成总体积50 μl的pcr扩增体系,pcr程序为98℃预变性45 sec,98℃变性15 sec,60℃退火30 sec,72℃延伸30 sec,扩增9 cycles,72℃完全延伸1 min,4℃ hold,热盖105℃;(11)向扩增产物中加入1.8
×
的纯化磁珠,充分混匀后室温孵育5 min,上磁力架至液体澄清,弃上清,加入200 μl 80%乙醇,1min后弃上清,重复清洗一次,在室温下将磁珠混合物干燥4 min,晾干后用32 μl水重悬,室温下孵育5 min,上磁力架至液体澄清,转移30 μl上清液,即为探针捕获文库,并进行qubit定量和2100质检,质控合格后,采用illumina测序平台测序仪进行上机测序,测序实验操作按照制造商提供的操作说明书进行上机测序操作。
46.实施例4本实施例对hrr基因子探针组与snp位点子探针组的不同混合比例进行测试,按实施例3所述,对相同样本文库分别按摩尔浓度比为2:1、3:1、4:1、5:1和10:1共计5种比例混合本发明hrr基因子探针组与snp位点子探针组,并进行相同数据量测序,分析结果如图4所示,随着hrr基因子探针组与snp位点子探针组的相对摩尔浓度比例升高,hrr基因区的平均
覆盖深度增加,同时snp区的平均深度降低,但升降幅度不同,这是由于两部分的探针覆盖区域大小不同所导致的,可见本发明控制hrr基因子探针组与snp位点子探针组的摩尔浓度比为(2~5):1能够进一步控制hrr基因测序深度和snp位点测序深度平衡,当探针比例为4:1时,hrr基因测序深度和snp位点测序深度达到最佳平衡,即40个hrr基因区有效捕获深度可达到700
×
,snp区有效捕获深度可达到200
×
,两部分均能实现高深度的捕获,满足分析需求。
47.实施例5本实施例利用本发明的探针组合物对包含brca1/2基因点突变的外购标准品brca somatic multiplex i(gdna)(购自horizon discovery,货号hd795)按实施例2、3所述方法进行检测并对测序数据进行分析,分别将该标准品携带的突变位点稀释至突变频率为5%、2.5%及1%,每种稀释频率重复三次,以验证本发明对低频hrr基因突变的检测准确度、灵敏性及重复性,结果如表2所示。
48.表2由表2可知,利用本发明的探针组合物可检测突变频率低至1%的hrr基因突变,且实测频率与预期频率具有高度的一致性,具有良好的重复性,说明所述试剂盒具有良好的灵敏度、准确性和重复性,具有实际应用的价值。
49.实施例6本实施例按实施例1
‑
3所述方法对30例来自临床合作单位的卵巢癌ffpe组织切片样本进行检测,并对测序数据进行质控、回帖到人类基因组,回帖得到的bam文件,分别进行hrr基因点突变分析及插入/缺失变异分析及基因组不稳定性分析。其中,hrr基因变异采用varscan软件,基因组不稳定性分析采用以下流程:1)对bam文件进行每个杂合子snp上的每个等位基因的等位型计数;2)统计snp探针区域的测序深度;3)采用r软件对每个snp的baf(b allele frequency,b等位基因频率)进行纠正校准,将校准后的baf和logr(log r ratio,对数比值)使用ascat进行基因组拷贝数变化和区域划分;4)将结果输入sequenza软件进行不同区域的基因组不稳定类型评估并分别统计基因组杂合性缺失、大片段迁移和端粒等位基因不平衡的数目。最终样本的基因组不稳定评分为上述三种基因组不稳定事件数目的加和,检测结果如下表3。
50.对表3数据分析可知,本发明能够同时检出hrr基因变异及基因组不稳定性评分,最终可结合本发明列出的hrd阳性判断标准,即具有brca1/2基因致病或疑似致病突变和/
或基因组不稳定评分≥43即为hrd阳性,以上30例卵巢癌样本中,有11例样本为brca1/2突变,占比33%,与文献报道类似,除该11例样本外,另外6例样本(t21033、t21352、t21439、t21401、t21459、t1810411)虽不具有brca1/2基因突变,但基因组不稳定性评分≥43,也可判断为hrd阳性,在该样本集中,hrd阳性样本共计17例,占比56.7%,也与文献报道类似,表明本发明可用于卵巢癌的hrd检测,有效扩大同源重组缺陷阳性的检出率。
51.表3综上所述,本发明筛选能够充分表征基因组不稳定性的snp位点并结合同源重组修复(hrr)基因构建基因库,并设计探针组合物,利用所述探针组合物能够对hrr基因的变异进行检测,同时对基因组不稳定性进行准确评估,并控制hrr基因探针和snp位点探针的混合比例,在有限的测序量条件下,使得hrr基因区与snp区获得不同程度的高深度覆盖,同
时满足基因变异及不稳定性评估的深度要求,能够有效扩大同源重组缺陷阳性的检出率,并降低检测成本,提高了检测经济效益。
52.申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。