1.本发明属于药物化学技术领域,尤其是涉及一种黄酮类端锚聚合酶2抑制剂及其制备方法和应用。
背景技术:
2.端锚聚合酶是一种多功能蛋白质翻译修饰酶,是多腺苷二磷酸核糖聚合酶家族的一员,其主要结构域为锚蛋白重复序列。该酶不仅可与端粒重复序列因子结合,还能与轴抑制蛋白羧基端的高度保守结构域结合,实现了对端粒酶的抑制作用,从而调节细胞外因子(wnt)/β
‑
联蛋白(β
‑
catenin)信号通路,以达抑制肿瘤细胞的增殖与生长。2009年nature报道的一种端锚聚合酶抑制剂xav939就是基于端锚聚合酶1/2的晶体结构直接合成的,其可通过稳定轴抑制蛋白的表达选择性阻滞wnt/β
‑
catenin信号通路的活化,从而限制肿瘤细胞的增殖与生长。近年来,端锚聚合酶2(tnks2)作为一种新的抗肿瘤药物靶点备受关注。针对tnks开发的药物研究越来越多,显示出抗肿瘤药物的巨大发展前景。黄酮类化合物是自然界中广泛存在并具有抗氧化特性的一大类化合物。另外,黄酮类衍生物已被证明可抑制端锚聚合酶2,在肺癌、前列腺癌、结直肠癌、胰腺癌和卵巢癌细胞中具有抗增殖特性。大部分已报道的黄酮类端锚聚合酶2抑制剂仅作用于端锚聚合酶2的烟酰胺结合位点。
技术实现要素:
3.为了寻找一种新的端锚聚合酶2抑制剂,本发明在于提供一种新型的黄酮类端锚聚合酶2抑制剂及其制备方法,以及其在用于制备抑制端锚聚合酶2的药物方面的应用。
4.同时作用于烟酰胺和腺苷结合位点的抑制剂具有更好的抑制活性和选择性。本发明基于计算机辅助药物合成设计的方法,新设计合成的黄酮类化合物具有较长的支链,能够同时占据端锚聚合酶2的烟酰胺和腺苷结合位点,进而提高化合物的抑制活性和选择性。
5.本发明中,利用计算机辅助药物设计的方法针对端锚聚合酶2的结构设计并且合成了多个结构新颖的端锚聚合酶2抑制剂,最终对其进行端锚聚合酶2抑制活性的测试,并且与已报道的端锚聚合酶2抑制剂gk
‑
007的抑制活性(gk
‑
007的ic
50
=0.035um)进行对比,发现所合成的多个端锚聚合酶2抑制剂具有较好的端锚聚合酶2抑制活性,尤其是其中优选的5个化合物的ic
50
分别为8.74um、3.54um、1.25um、0.89um、0.082um。
6.本发明的目的可以通过以下技术方案来实现:
7.本发明第一方面提供一种黄酮类端锚聚合酶2抑制剂,具有式i所示的结构:
[0008][0009]
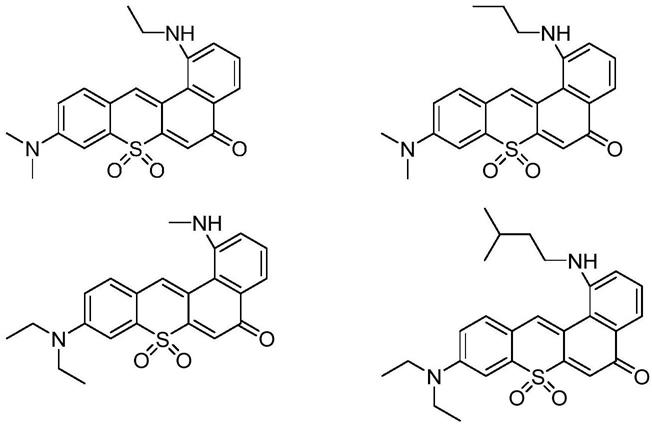
其中r选自:
[0010][0010]
中的任意一种。
[0011]
优选地,所述的r选自:
[0012]
中的任意一种。
[0013]
本发明第二方面提供所述的黄酮类端锚聚合酶2抑制剂的制备方法,该方法采用4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸和苯胺衍生物或苯甲酰肼衍生物作为起始原料,使用脲鎓盐作为缩合剂,在添加碱的条件下,于有机溶剂中进行缩合反应,获得所述的黄酮类端锚聚合酶2抑制剂;
[0014]
所述的苯胺衍生物为
[0015]
所述的苯甲酰肼衍生物为
[0016]
其中:
[0017]
r1选自h、ch3、och3、cl、br或3,4,5
‑
tri
‑
och3;
[0018]
r2选自h、ch3、och3、cl或br。
[0019]
反应方程式分别如下面a、b和c三个过程所示:
[0020][0021]
优选地,所述的脲鎓盐选自hatu、hbtu、hctu、tbtu、tstu或tntu。优选地,所述的4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸与缩合剂的摩尔比为1:1~1:5
[0022]
优选地,所述的4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸与苯胺衍生物或苯甲酰肼衍生物的摩尔比为1:1~1:3。
[0023]
优选地,所述的4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸与碱的摩尔比为1:1~1:5。
[0024]
优选地,所述的碱选自dipea或者三乙胺。
[0025]
优选地,有机溶剂选自dmso、dmf或nmp。
[0026]
优选地,缩合反应的反应温度为0℃~70℃。
[0027]
本发明第三方面提供所述的黄酮类端锚聚合酶2抑制剂在用于制备抑制端锚聚合酶2的药物方面的应用。
[0028]
与现有技术相比,本发明的有益效果在于:
[0029]
(1)本发明的制备方法简单、成本低、反应条件温和。
[0030]
(2)本发明合成的化合物结构新颖,均为首次报道。
[0031]
(3)本发明合成的黄酮类端锚聚合酶2抑制剂效果较好,其中化合物5(参见实施例5)具有和gk
‑
007相近的抑制活性以及更好的选择性。
附图说明
[0032]
图1为实施例1制得的产物的氢谱(1h nmr(dmso))。
[0033]
图2为实施例1制得的产物的碳谱(
13
c nmr(dmso))。
[0034]
图3为实施例2制得的产物的氢谱(1h nmr(dmso))。
[0035]
图4为实施例2制得的产物的碳谱(
13
c nmr(dmso))。
[0036]
图5为实施例3制得的产物的氢谱(1h nmr(dmso))。
[0037]
图6为实施例3制得的产物的碳谱(
13
c nmr(dmso))。
[0038]
图7为实施例4制得的产物的氢谱(1h nmr(dmso))。
[0039]
图8为实施例4制得的产物的碳谱(
13
c nmr(dmso))。
[0040]
图9为实施例5制得的产物的氢谱(1h nmr(dmso))。
[0041]
图10为实施例5制得的产物的碳谱(
13
c nmr(dmso))。
具体实施方式
[0042]
一种黄酮类端锚聚合酶2抑制剂,具有式i所示的结构:
[0043][0044]
其中r选自:
[0045][0045]
中的任意一种。
[0046]
优选r选自:
[0047]
中的任意一种。
[0048]
所述的黄酮类端锚聚合酶2抑制剂的制备方法,采用4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸和苯胺衍生物或苯甲酰肼衍生物作为起始原料,使用脲鎓盐作为缩合剂,在添加碱的条件下,于有机溶剂中进行缩合反应,获得所述的黄酮类端锚聚合酶2抑制剂;
[0049]
所述的苯胺衍生物为
[0050]
所述的苯甲酰肼衍生物为
[0051]
其中:
[0052]
r1选自h、ch3、och3、cl、br或3,4,5
‑
tri
‑
och3;
[0053]
r2选自h、ch3、och3、cl或br。
[0054]
优选地脲鎓盐选自hatu、hbtu、hctu、tbtu、tstu或tntu。优选4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸与缩合剂的摩尔比为1:1~1:5。优选4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸与苯胺衍生物或苯甲酰肼衍生物的摩尔比为1:1~1:3。优选4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸与碱的摩尔比为1:1~1:5。优选碱选自dipea或者三乙胺。优选有机溶剂选自dmso、dmf或nmp。优选缩
合反应的反应温度为0℃~70℃。
[0055]
本发明第三方面提供所述的黄酮类端锚聚合酶2抑制剂在用于制备抑制端锚聚合酶2的药物方面的应用。
[0056]
下面结合附图和具体实施例对本发明进行详细说明。
[0057]
本发明中,ic
50
值的测试方法:化合物对tnks2的抑制活性通过酶促反应测定。将待测化合物先溶解于dmso溶液中,配制成初始浓度为200μm/l,然后按比例每次稀释5倍,配置7个浓度梯度,得到每个待测化合物的溶液浓度范围为0.0128μm/l到200μm/l。酶促反应在室温下在96孔板(greiner bio
‑
one u形黑板)中进行。将5nm的tnks2在测定缓冲液(50mm hepes ph 7.0、1mm chaps)中与化合物和500nm nad'一起孵育1小时,每个浓度梯度同时设置四个平行实验组。然后加入检测缓冲液(0.1%bsa的50mm hepes,0.8m kf和20mm edta)在室温反应1小时。最后加入20ml 20%苯乙酮的乙醇溶液和20ml 2mkoh终止反应,未反应的nad'被化学转化为荧光物质,在激发波长355nm,吸收波长450nm条件下读取荧光强度。按照公式:抑制率=(样品荧光强度
‑
空白)/(酶值荧光强度
‑
空白)计算每个抑制剂相应的抑制率,然后使用graph pad prism软件拟合出半数有效抑制浓度。
[0058]
实施例1
[0059]
n
‑
(4
‑
乙酰苯基)
‑4‑
氧代
‑
4h
‑
苯并吡喃
‑2‑
甲酰胺
[0060][0061]
将4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸(95mg,0.5mmol)溶于25ml dmf中,冰浴条件下,边搅拌边加入hatu(228mg,0.6mmol)和dipea(129mg,1mmol)至含有4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸的dmf溶液中,缓慢升温至室温,15分钟后加入4
‑
氨基苯乙酮(81mg,0.6mmol)。室温下继续搅拌6h,直至4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸反应完全。反应完成后,将反应液倒入冰水中。析出固体,收集固体并用水、10%na2co3、盐水和乙醇超声洗涤。固体经快速硅胶色谱法(石油醚/乙酸乙酯=6:1)提纯得到产物。
[0062]
ic
50
值为8.74um。
[0063]1h nmr(400mhz,dmso)δ=10.94(s,1h),8.06(d,j=8.0hz,1h),7.98(q,j=8.6hz,4h),7.90(d,j=7.6hz,1h),7.83(d,j=8.5hz,1h),7.55(t,j=7.6hz,1h),6.98(s,1h),2.55(s,3h).
13
c nmr(101mhz,dmso)δ=196.73,177.32,158.20,155.34,155.20,141.93,135.15,133.12,129.38,126.23,125.02,123.78,120.36,119.09,111.43,26.59.
[0064]
氢谱和碳谱参见图1和图2。
[0065]
实施例2
[0066]
n'
‑
苯甲酰基
‑4‑
氧代
‑
4h
‑
苯并吡喃
‑2‑
碳酰肼
[0067][0068]
将4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸(95mg,0.5mmol)溶于25ml dmf中,冰浴条件下,
边搅拌边加入hatu(228mg,0.6mmol)和dipea(129mg,1mmol)至含有4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸的dmf溶液中,缓慢升温至室温,15分钟后加入苯甲酰肼(82mg,0.6mmol)。室温下继续搅拌6h,直至4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸反应完全。反应完成后,将反应液倒入冰水中。析出固体,收集固体并用水、10%na2co3、盐水和乙醇超声洗涤。固体经快速硅胶色谱法(石油醚/乙酸乙酯=4:1)提纯得到产物。
[0069]
ic
50
值为3.54um。
[0070]1h nmr(400mhz,dmso)δ11.17(s,1h),10.75(s,1h),8.07(t,j=7.5hz,1h),7.92(t,j=7.6hz,3h),7.76(d,j=7.9hz,1h),7.57(dt,j=24.0,7.6hz,4h),6.90(d,j=6.7hz,1h).
13
c nmr(101mhz,dmso)δ=177.13,165.76,158.73,155.18,154.73,135.28,132.27,132.18,128.68,127.57,126.27,125.07,123.83,118.90,111.32.
[0071]
氢谱和碳谱参见图3和图4。
[0072]
实施例3
[0073]
n'
‑
(4
‑
甲基苯甲酰基)
‑4‑
氧代
‑
4h
‑
苯并吡喃
‑2‑
碳酰肼
[0074][0075]
将4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸(95mg,0.5mmol)溶于25ml dmf中,冰浴条件下,边搅拌边加入hatu(228mg,0.6mmol)和dipea(129mg,1mmol)至含有4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸的dmf溶液中,缓慢升温至室温,15分钟后加入4
‑
甲基苯甲酰肼(90mg,0.6mmol)。室温下继续搅拌6h,直至4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸反应完全。反应完成后,将反应液倒入冰水中。析出固体,收集固体并用水、10%na2co3、盐水和乙醇超声洗涤。固体经快速硅胶色谱法(石油醚/乙酸乙酯=4:1)提纯得到产物。
[0076]
ic
50
值为1.25um。
[0077]1h nmr(400mhz,dmso)δ=8.07(d,j=8.0hz,1h),7.91(t,j=7.8hz,1h),7.83(d,j=7.8hz,2h),7.75(d,j=8.5hz,1h),7.56(t,j=7.5hz,1h),7.33(d,j=7.8hz,2h),6.90(s,1h),2.38(s,3h).
13
c nmr(101mhz,dmso)δ177.18,165.60,158.72,155.22,154.88,142.24,135.30,129.49,129.20,127.63,126.27,125.07,123.85,118.93,111.29,21.12.
[0078]
氢谱和碳谱参见图5和图6。
[0079]
实施例4
[0080]
n'
‑
(4
‑
氯苯甲酰基)
‑4‑
氧代
‑
4h
‑
苯并吡喃
‑2‑
碳酰肼
[0081][0082]
将4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸(95mg,0.5mmol)溶于25ml dmf中,冰浴条件下,边搅拌边加入hatu(228mg,0.6mmol)和dipea(129mg,1mmol)至含有4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸的dmf溶液中,缓慢升温至室温,15分钟后加入4
‑
氯苯甲酰肼(102mg,0.6mmol)。室温下继续搅拌6h,直至4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸反应完全。反应完成后,将反应液
倒入冰水中。析出固体,收集固体并用水、10%na2co3、盐水和乙醇超声洗涤。固体经快速硅胶色谱法(石油醚/乙酸乙酯=4:1)提纯得到产物。
[0083]
ic
50
值为0.89um。
[0084]1h nmr(400mhz,dmso)δ11.05(s,3h),8.10(d,j=7.9hz,1h),7.95(dd,j=15.1,7.9hz,3h),7.78(d,j=8.5hz,1h),7.65(d,j=8.1hz,2h),7.59(t,j=7.6hz,1h),6.94(s,1h).
13
c nmr(101mhz,dmso)δ=177.14,164.79,158.73,155.19,154.67,137.09,135.30,130.97,129.52,128.86,126.29,125.09,123.85,118.90,111.37.
[0085]
氢谱和碳谱参见图7和图8。
[0086]
实施例5
[0087]4‑
氧代
‑
n
‑
(4
‑
(3,4,5
‑
三甲氧基苯甲酰胺基)苯基)
‑
4h
‑
苯并吡喃
‑2‑
甲酰胺
[0088][0089]
将4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸(95mg,0.5mmol)溶于25ml dmf中,冰浴条件下,边搅拌边加入hatu(228mg,0.6mmol)和dipea(129mg,1mmol)至含有4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸的dmf溶液中,缓慢升温至室温,15分钟后加入n
‑
(4
‑
氨基苯基)
‑
3,4,5
‑
三甲氧基苯甲酰胺(181mg,0.6mmol)。室温下继续搅拌6h,直至4
‑
氧代
‑
4h
‑1‑
苯并吡喃
‑2‑
羧酸反应完全。反应完成后,将反应液倒入冰水中。析出固体,收集固体并用水、10%na2co3、盐水和乙醇超声洗涤。固体经快速硅胶色谱法(石油醚/乙酸乙酯=4:1)提纯得到产物。
[0090]
ic
50
值为0.082um。
[0091]1h nmr(400mhz,dmso)δ10.73(s,1h),10.17(s,1h),8.07(d,j=7.7hz,1h),7.93
–
7.87(m,1h),7.83(d,j=8.6hz,1h),7.79(s,4h),7.54(t,j=7.5hz,1h),7.28(s,2h),6.97(s,1h),3.87(s,8h),3.73(s,3h).
13
c nmr(101mhz,dmso)δ=177.39,164.87,157.55,155.83,155.24,152.71,140.41,136.04,135.09,133.33,130.04,126.18,125.02,123.79,121.50,121.04,119.09,111.08,105.38,60.21,56.20.
[0092]
氢谱和碳谱参见图9和图10。
[0093]
上述对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。